
1. Предмет і завдання патофізіології, зв’язок з іншими науками, значення для клініки. Патофізіологія як навчальна дисципліна.
Патологічна фізіологія - це наука, що вивчає загальні закономірності виникнення, розвитку та завершення хвороби. Це наука про життєдіяльність хворого організму.
Зв'язок патологічної фізіології з іншими науками:
1. Зв'язок з науками, що вивчають властивості чинників навколишнього середовища, здатних викликати хвороби (фізика, хімія, біологія, мікробіологія, соціологія). Ці науки дають відомості, необхідні для вивчення етіології.
2.Зв'язок з науками, що вивчають властивості організму і його життєдіяльність (цитологія, ембріологія, гістологія, нормальна фізіологія, біохімія, імунологія, генетика). Ці науки створюють основу для вивчення патогенезу.
3.Зв'язок з загальнотеоретичними науками, що вивчають хворобу (патологічна анатомія, фармакологія). Ці науки разом з патологічною фізіологією створюют ьцілісну картину хвороби.
4.Зв'язок з клінічними науками. Патологічна фізіологія визначає основні етіологічні і патогенетичн іпринципи профілактики, діагностики і лікування хвороби.
Завдання дисципліни:
1.З'ясування сутності хвороби (що таке хвороба?).
2.Вивчення причин і умов виникнення хвороби.
3.Розкриття механізмів розвитку хвороби і окремих її проявів, установлення закономірностей перебігу хвороби і механізмів видужання.
4.Визначення загальних принципів профілактики і лікування хвороб.
Розв'язання зазначених завдань відбувається в рамках чотирьох складовихчастин патологічної фізіології як науки: нозології, етіології, патогенезу, експериментальної терапії.
Відповідно до завдань, які повинна розглядати патофізіологія як фундаментальна наука, вона як найважливіший розділ біології та медицини має три складові частини:
• нозологія (вчення про хворобу);
• типові патофізіологічні процеси;
• патофізіологія органів і систем.
Клініка порушує перед патофізіологами проблеми і надає необхідний для їх розв'язання фактичний матеріал.
Патофізіологія, будучи науковою основою медицини (філософією медицини), установлює етіологію і патогенез хвороб і на цій основі визначає головні напрями їх попередження і лікування
Складові частини патологічної фізіології як навчальної дисципліни.
1. Загальна патологія.
2. Патофізіологія органів і систем.
Загальна патологія охоплює широке коло питань, об'єднаних у розділи: нозологія, патогенна дія факторів зовнішнього середовища, роль внутрішніх чинників у патології, типові патологічні процеси, типові порушення обміну речовин.
Патофізіологія органів і систем вивчаєз агальні закономірност ірозвитку патологічних процесів в окремих функціональних системах, а також етіологію і патогенез найпоширеніших нозологічних форм.
2. Методи патофізіології. Експеримент, його значення для вирішення фундаментальних проблем медицини. Види експерименту. Основні етапи проведення експериментальних досліджень. Сучасні методи і методики моделювання патологічних процесів.
Для вивчення хвороби використовують - клінічний, епідеміологічний, анатомічний, експериментальний методи.
Клінічний метод передбачає вивчення хвороби безпосередньо біля ліжка хворої людини. Патофізіологія завжди використовувала цей метод для розв'язання поставлених перед нею завдань. Сучасні досягнення науки уможливлюють дослідження великої кількості функціональних, біохімічних, імунологічних та інших показників у хворої людини. У зв'язку із цим сьогодні досить швидко розвивається один з напрямів патологічної фізіології - клінічна патофізіологія.
Епідеміологічний метод має об'єктом вивчення хвороби популяцію людей. Він відіграє важливу роль при встановленні причин виникнення і закономірностей розвитку інфекційних захворювань. Патофізіологія не застосовує цей метод безпосередньо, але використовує отримані з його допомогою дані, що стосуються етіології основних неінфекційних хвороб (наприклад, вивчення факторів ризику атеросклерозу).
Анатомічний метод є основним методом патологічної анатомії, яка разом з патофізіологією вивчає сутність хвороби, використовуючи при цьому як об'єкт досліджень труп людини.
Експериментальний метод передбачає вивчення хвороби на лабораторних тваринах, які єоб'єктом моделювання хвороб людини.
Основний метод патофізіології- експеримент. Експеримент — це активний вплив людини на природу і штучне відтворення різних її явищ з метою пізнання об'єктивних закономірностей. Особливість патофізіологічного експерименту полягає у відтворенні на лабораторних тваринах експериментальних моделей хвороб з метою встановлення механізмів їх виникнення, розвитку і завершення у людини.
Хоча експеримент на тваринах не відтворює повну картину відповідної хвороби у людини, він дає можливість спостерігати і вивчати хворобу від самого початку до її завершення, що неможливо в клініці. В експерименті можна контролювати умови навколишнього середовища, що впливають на перебіг хвороби, а також вихідний стан тварин, які є об'єктом досліду. Експеримент - це єдина можливість вивчення таких впливів на організм, використання яких у клініці неприпустимо (травма, трансплантація пухлин, опромінення). Експериментальні дослідження дають об'єктивний матеріал для побудови наукових теорій.
Види експерименту:
Гострий експеримент (вівісекція) ґрунтується на оперативному втручанні в організм тварини. Він дозволяє вивчати гострі розлади в організмі, наприклад шок, колапс, крашсиндром, гостру недостатність дихання, кровообігу, нирок та ін.
Хронічний експеримент — тривалий, дає можливість вивчати динаміку розвитку хвороби. Його використовують для моделювання хронічних хвороб, наприклад атеросклерозу, артеріальної гіпертензії, цукрового діабету, виразкової хвороби та ін.
Етапи проведення патофізіологічного експерименту:
1.Планування експерименту.
2.Моделювання патологічного процесу.
3.Отримання інформації про зміни в організмі експериментальних тварин.
4.Аналіз і синтез отриманих результатів.
Методи моделювання хвороб:
Метод видалення. Видаленням печінки моделюють печінкову недостатність, нирок — гостру ниркову недостатність, підшлункової залози — цукровий діабет, гіпофіза - пангіпопітуїтаризм.
Метод руйнування (ушкодження). Із цією метою використовують хірургічні втручання (перетинання нервів, руйнування нервових центрів, ушкодження тканин), фізичні фактори (іонізуюча радіація, температура та ін.), хімічні агенти (отрути, інгібітори), імунні впливи (протитканинні сироватки, антитіла).
Метод перевантажень. Викликаючи функціональні перевантаження вищих центрів ЦНС, моделюють неврози; створюючи штучні вади серця, відтворюють серцеву недостатність; уведенням великих кількостей холестеролу й хлориду натрію моделюють відповідно атеросклероз і артеріальку гіпертензію.
Метод створення дефіциту. Створюючи дефіцит кисню в барокамері, моделюють гіпоксію; застосовуючи раціони, що не містять вітамінів, отримують а- і гіповітамінози, антиоксидантну недостатність.
Метод порушення нервової і гормональної регуляції. Широко використовують подразнення або ушкодження нервових структур, фармакологічні втручання в обмін нейромедіаторів, уведення великих кількостей гормонів або їхніх аналогів.
Метод створення перешкод. Перев'язуючи кровоносні судини, моделюють інфаркт міокарда і артеріальну гіпертензію, а загальну жовчну протоку — механічну жовтяницю. Введення в коронарні судини емболів використовують для відтворення інфаркту міокарда.
Метод екзогенної індукції. Вводять в організм або діють на нього факторами, які є специфічними збудниками хвороби. Так моделюють усі інфекційні хвороби, злоякісні пухлини, алергію.
Метод трансплантації застосовують при вивченні злоякісних пухлин, склеротичних уражень судин.
Метод експлантації - вивчення патологічних процесів поза організмом: в культурах тканин, на ізольованих органах. Використовують при вивченні пухлинного росту, атеросклерозу, алергічних реакцій.
3. Основні поняття нозології: здоров’я, хвороба, патологічний стан, патологічний процес, типовий патологічний процес, патологічна реакція.
Основні поняття загальної нозології: здоров'я, норма, хвороба, патологічний процес, патологічний стан, патологічні реакції.
Здоров'я - це стан повного фізичного, психологічного й соціального благополуччя, а не тільки відсутність хвороб чи фізичних вад (визначення ВООЗ, 1946); це біологічний оптимум функціонування і розвитку організму.
Хвороба - це порушення нормальної життєдіяльності організму внаслідок впливу на нього шкідливих агентів, що спричинює зниження його пристосувальних можливостей, працездатності й збільшує ймовірність смерті (М. Н.Зайко).
Патологічний стан — це сукупність патологічних змін в організмі, що виникають унаслідок розвитку патологічного процесу (стійке відхилення від норми, що має біологічно негативні наслідки для організму)
Патологічний процес - це певна послідовність змін і реакцій, що закономірно виникають і розвиваються в організмі внаслідок дії патогенного фактора. Цепоєднанняпроцесівруйнування (ушкодження) і захиснихкомпенсаторнихреакцій, щовиникають на різнихрівняхорганізаціїорганізмупіддією патогенного фактора.
Типові патологічні процеси - це процеси, які розвиваються за однаковими законами, незалежно від причини, локалізації, виду тварин та індивідуальних особливостей організму. Прикладами таких процесів можуть бути запалення, пухлинний процес, місцеві розлади кровообігу, гіпоксія, голодування, гарячка.
Патологічна реакція— це неадекватна і біологічно недоцільна відповідь організму чи його систем на дію звичайних або надмірних подразників. Патологічна реакція є елементом руйнівного, власне патологічного боку патологічного процесу. Приклади: алергія, патологічні рефлекси.
4. Основні напрями вчення про хворобу (гуморальний, солідарний, целюлярний), їх сучасний стан.
Гуморальна патологія — це система теоретичних поглядів, згідно з якими основним механізмом розвитку хвороб є порушення стану рідин організму. Основоположником цього напряму є Гіппократ. Його послідовниками були Гален, Ібн-Сіна (Авіценна), Парацельс, Рокитан-ський, Сельє.
На сучасному етапі гуморальний напрям відображається у розв'язанні таких проблем, як порушення гуморальних механізмів регуляції (ендокринологія), роль гуморальних факторів у розвитку патологічних процесів (медіатори запалення, регулятори проліферації та ін.), гуморальні механізми імунітету.
Солідарний напрям — це система теоретичних поглядів, згідно з якими сутність хвороби полягає в порушенні співвідношення і характеру руху твердих часток організму (sol-idus - твердий). Цей напрям виник на основі атомістичного вчення Демокрита. Його основоположником вважається давньогрецький лікар Асклепіад (II—І ст. до н.е.).
Сучасним етапом розвитку цього напряму варто вважати молекулярну патологію, що вивчає молекулярні механізми розвитку хвороб (спадкова патологія, молекулярна онкологія, імуногенетика та ін.).
Клітинна (целюлярна) патологія — це напрям, що розглядає клітину як матеріальний субстрат хвороби, а саму хворобу — як певну суму уражень великої кількості окремих клітин.
Основні положення цієї теорії було сформульовано в роботах Р. Вірхова в 1855-1858 pp. У вигляді таких тверджень:
1.Хвороба організму - це хвороба його клітин.
2.Хвороба завжди має свою локалізацію.
3.Хвороба виникає хоча б за однієї з трьох умов (тріада Вірхова); а) гетерометрія, тобто зміна кількості клітин; б) гетеротопія — поява клітин не на своєму місці; в) гетерохронія — поява клітин не у свій час.
5. Філософські, медико-біологічні та соціальні аспекти вчення про хворобу. Принципи класифікації хвороб.
Існує кілька принципів класифікації хвороб:
1.Етіологічний, відповідно до якого виділяють спадкові і набуті, інфекційні і неінфекційні хвороби.
2. Анатомо-топографічний - серцево-судинні хвороби, хвороби органів дихання, хвороби нирок та ін.
3.За віком і статтю — дитячі хвороби, хвороби старечого віку, жіночі хвороби.
4.Екологічний — тропічні хвороби, хвороби Крайньої Півночі та ін.
5.Залежно від рівня уражень - молекулярні хвороби, хромосомні хвороби.
6.Соціальний - професійні хвороби, хвороби воєнного часу, "хвороби цивілізації'
7.Патогенетичний принцип - алергічні, запальні, пухлинні, обмінні та інші хвороби.
8.Залежно від співвідношення структурних і функціональних порушень — органічні і функціональні хвороби
9.За клінічним перебігом - гострі, підгострі і хронічні.
10.Залежно від методів, які переважно використовують для лікування хвороб, - терапевтичні і хірургічні хвороби.
Чим відрізняється хвороба відздоров'я:
1. Немає якісних відмінностей між хворобою і здоров'ям. Інакше кажучи, хвороба відрізняється від здоров'я тільки кількісними характеристиками (К. Бернар, Р. Вірхов, І. В. Давидовський).
2. У хворобі виникає нова якість, однак вона не є результатом появи чогось принципово нового, а є наслідком переходу кількісних змін у якісні (О. О. Богомолець).
3. Хвороба – це якісноновий стан організму. Існують закони розвитку хвороби, які багато в чому відрізняються від законів життєдіяльності організму (В. В. Пашутін).
6. Універсальні періоди в розвитку хвороби. Варіанти завершення хвороб. Поняття про термінальні стани: агонія, клінічна смерть, біологічна смерть. Принципи реанімації.
У розвитку (перебігу) хвороби розрізняють такі чотири періоди:
1) прихований (латентний);
2) продромальний (період передвісників);
3) період розпалу (виражених клінічний ознак хвороби);
4) завершальний (реконвалесцентний, видужання).
Закінчення хвороби може бути різним, а саме:
-повне одужання;
-неповне одужання;
-перехід у патологічний стан;
-рецидив;
-ремісія;
-термінальний стан;
-смерть.
Принципи реанімації:
Спочатку необхідно припинити вплив шкідливих чинників, наприклад: перервати контакт з електричною напругою, вивільнити повішеного з петлі, погасити палаючий одяг, відновити прохідність дихальних шляхів.
Хворого треба укласти горизонтально на тверду поверхню (підлога, земля, щит) і звільнити груднину від одягу, що стискує.
Починати реанімацію необхідно з штучної вентиляції легень (ШВЛ) методом «з рота в рот» або «з рота в ніс» і закритого масажу серця.
ШВЛ «з рота в ніс» фізіологічніше, ніж ШВЛ «з рота в рот». Але частіше проводиться остання через виявлення у помираючих аденоїдів, викривленої носової перегородки і затруднення механічного очищення носа. Однак, коли у них виникають судомні скорочення жувальних м'язів і неможливо відкрити рот або утворюється перешкоду під час роздування легень методом «з рота в рот», необхідно проводити ШВЛ «з рота в ніс».
Щоб ШВЛ була ефективною, попередньо необхідно виконати такі умови: Насамперед звільнити верхні дихальні шляхи;
Забезпечити адекватний для певного віку дихальний об'єм і герметичність губ.
Верхні дихальні шляхи звільняють так:
підводять одну руку під шию, а іншою, покладену на лоб, помірно закидають голову;
розкривають рот, якщо він залишився закритим після закидання голови; необхідно руку з-під шиї змістити на підборіддя, обережно його підтримуючи і відтягуючи нижню губу;
висувають вперед нижню щелепу, захопивши II—IV пальцями обох рук її висхідні гілки біля вушної раковини і зміщуючи таким чином, щоб нижні зуби виявилися попереду верхніх або в одній площині з ними: нижня губа зміщується великими пальцями;
поєднують помірне закидання голови з висуванням нижньої щелепи;
механічно очищають ротову порожнину від слизу, блювотних мас, сторонніх тіл, зубних протезів.
При виконанні ШВЛ «з рота в ніс» після звільнення верхніх дихальних шляхів однією рукою піднімають підборіддя хворого, щоб закрити його рот, зробивши помірно глибокий вдих, щільно притискають свої губи до носа потерпілого, в який вдувають повітря. Під час видиху завжди необхідно відкривати рот реанімованого для вилучення можливого перешкоди вдиху повітря.
Агонія (грец. αγωνια — боротьба) — сукупність останніх проявів життєвих функцій, стан / синдром, що безпосередньо передує смерті. Під час агонії глибоко порушується діяльність вищих відділів головного мозку, затьмарюється свідомість, згасає діяльність усіх органів чуття, різко падає серцева діяльність. Закінчується агонія з останнім ударом серця.
Клінічна смерть — це такий стан організму, який виникає протягом декількох хвилин від 3 до 5 хвилин після зупинки дихання та кровообігу, коли зникають всі зовнішні прояви життєдіяльності
7. Визначення поняття “етіологія”. Основні напрями вчення про етіологію: монокаузалізм, кондиціоналізм, конституціоналізм, психосоматична концепція. Проблема причинності в патології, сучасний стан її вирішення.
Етіоло́гія в медицині (грец. αἰτία — причина та грец. λόγος — думати) — теоретичний розділ медицини, який вивчає причини виникнення хвороб, зокрема інфекційних. Також часто це у вузькому розумінні визначають як окреслення безпосередньої причини хвороби, синдрому або патологічного стану.

8. Класифікація етіологічних чинників, поняття про фактори ризику. „Хвороби цивілізації”.
Виділяють такі причини хвороб:
Механічні (закриті та відкриті травми, струс, тощо).
Фізичні (висока або низька температура, електричний струм, тощо).
Хімічні (промислові та побутові токсичні речовини, тощо).
Біологічні (дія патогенних бактерій, найпростіших, вірусів, грибків, пріонів, мікробних токсинів, тощо).
Психогенні, включаючи соціальні (війни, дискримінація, урбанізація, тощо).
Генетичні (спадкові).
Хворобами цивілізації називають атеросклероз, цукровий діабет II типу (інсулінонезалежний), гіпертонічну хворобу, а також нерідко ожиріння. Протягом останніх десятиліть ці хвороби помолодшали й часто трапляються вже в людей після 30 років. За даними деяких дослідників, атеросклероз аорти і коронарних артерій є практично в кожного, з віком лише посилюється його прояв. Таке масове поширення, по суті — пандемія судинної патології, дозволяє припускати, що в умовах цивілізації людство до цієї патології схильне. При цьому, природно, рівень схильності в різних людей неоднаковий.
9. Принципи етіотропної, патогенетичної і симптоматичної терапії.
Суть етіотропної терапії (aetiotropus: від грец. - aetia - причина та tropos - повертання, подіяти на причину) полягає у застосуванні засобів, які діють на причину хвороби. Вона застосовується у тих випадках, коли етіологічний фактор знаходиться в організмі і продовжує впливати на нього
Патогенетична терапія
Терапія, що спрямована на механізми розвитку патологічного процесу, відновлення функцій органів і тканин, нормалізацію складу внутрішнього середовища шляхом впливу на нервову та ендокринну системи, підвищення неспецифічної резистентності
Симптоматична терапія - застосування засобів терапії, спрямова них на ліквідацію або послаблення найбільш загрозливих для життя симптомів хвороби
10. Визначення поняття “патогенез”. Патологічні (руйнівні) і пристосувально - компенсаторні (захисні) явища в патогенезі (на прикладах гострої променевої хвороби, запалення, крововтрати).
Патогене́з (від грец. páthos — страждання, хвороба і génesis — походження, виникнення) — механізми виникнення і розвитку хвороби і окремих її проявів на різних рівнях організму — від молекулярних порушень до змін в органах і системах
11.Причинно-наслідкові зв’язки, роль circulus vitiosus в патогенезі; поняття про головну та побічні ланки патогенезу. Специфічні та неспецифічні механізми розвитку хвороб.
У патогенезі хвороби можна виділити ряд етапів або ланок, які зв'язані між собою причинно-наслідковими зв'язками. Наприклад, перша ланка в патогенезі травматичного шоку — біль. Сильний біль призводить до пригнічення життєво важливих центрів, зокрема до зниження артеріального тиску, що є причиною кисневого голодування. Гіпоксія головного мозку приводить до ще більшого пригнічення судиннорухового центру й ще більшому зниженню артеріального тиску. Як видно, зміна причин і наслідків призвела до утворення порочного кола (circulus vitiosus). Значення порочного кола можна проілюструвати також на прикладі метеоризму. Здуття кишок гальмує їх моторну і секреторну функції, а це сприяє бродінню, утворенню газів і ще більшому метеоризму.
Специфічні та неспецифічні механізми розвитку хвороб. У розвитку хвороби зазвичай розрізняють чотири періоди (стадії): латентний, продромальний, період розпалу хвороби і результат, або період закінчення хвороби. Така періодизація склалася у минулому при клінічному аналізі гострих інфекційних хвороб (черевний тиф, скарлатина і т. д.). Інші хвороби (серцево-судинні, ендокринні, пухлини) розвиваються по інших закономірностях, і тому приведена періодизація до них мало застосовна. А. Д. Адо виділяє три стадії розвитку хвороби: початок, стадію власне хвороби, результат.
12.Поняття про недоношеність та затримку росту плода: етіологічні чинники та наслідки для дитини
Недоношений новонароджений –
Недоношеним вважається новонароджений, що народився між 28-м та 38-м тижнем вагітності з масою тіла менше 2500 г та довжиною тіла менше 45 см.
Недоношеність
І ступеня – 35-37 тижнів;
ІІ ступеня – 32-34 тижні;
ІІІ ступеня – 29-31 тиждень (глибоконедоношені);
ІV ступеня – 28 тижнів і менше (екстремально
недоношені).
Оновні фактори ризику, що збільшують смертність недоношених дітей, наступні:
1. Кровотеча у матері перед пологами;
2. Багатоплідна вагітність;
3. Пологи в тазовому передлежанні;
4. Відсутність стероїдної терапії у матері
для профілактики СДР;
5. Перинатальна асфіксія;
6. Чоловіча стать;
7. Гіпотермія;
8. Хвороба гіалінових мембран.
Основні проблеми новонароджених з малою масою тіла при
народженні
|
|
Недоношений новонароджений |
Новонароджений з затримкою внутрішньоутробного розвитку |
|
1 |
Респіраторний дистрес-синдром |
1. Гіпотермія |
|
2. |
Гіпоглікемія |
2. Гіпоглікемія |
|
3. |
Гіпотермія |
3. Проблеми з вигодовуванням |
|
4. |
Проблеми з вигодовуванням |
4. Гіпербілірубінемія |
|
5. |
Гіпербілірубінемія |
5. Інфекції |
|
6. |
Інфекції |
6. Поліцитемія |
|
7. |
Апное |
7. Природжені аномалії розвитку 8. Гіпокальціемія |
|
8. |
Гіпокальціемія |
|
|
9. |
Анемія |
|
Затримка росту плода (ЗРП) - ускладнення вагітності, яке розвивається внаслідок плацентарної недостатності і призводить до народження дитини з масо-ростовими параметрами нижче 10-ої перцентилі для даного терміну вагітності.
Фактори ризику ЗРП:
1. Медичні:
- хронічна артеріальна гіпертензія;
- цукровий діабет;
- системні захворювання сполучної тканини;
- тромбофілії;
- захворювання нирок;
- прееклампсія вагітних;
- багатоплідна вагітність;
- крововтрата під час вагітності;
- аномалії пуповини та розташування плаценти;
- перинатальні інфекції;
- ЗРП в анамнезі;
- хромосомні та генетичні порушення;
- медикаменти (варфарин, фенітоїн).
2. Соціально-економічні:
- недостатнє харчування;
- тютюнопаління, вживання алкоголю, наркотиків;
- забруднення навколишнього середовища;
- професійні шкідливості.
Причина затримки росту може бути пов'язана з материнськими проблемами (підвищений тиск, цукровий діабет), а також плодовими факторами (пороки розвитку, хромосомні аномалії) або плацентарних патологіями.
13. Закономірності розвитку механічної травми. Травматичний шок. Синдром тривалого розчавлення.
Патогенна дія факторів зовнішнього середовища пов‘язана, головним чином, із впливом фізичних чинників: 1) механічної травми, 2) високої і низької температури, 3) іонізуючого випромінювання, 4) високого і низького атмосферного тиску, 5) електричного струму, 6) ультрачервоного і ультрафіолетового випромінювання.
Механічна травма - це ушкодження тканини твердим тілом чи поширенням вибухової хвилі. Місцево травма проявляється у вигляді розривів, забитих місць, переломів, роздавлювання чи їхньою комбінацією. Часто механічна травма поєднується з крововтратою, ушкодженням нервових стовбурів і шкірних покривів. Найбільш важкими загальними проявами травми є травматичний шок і синдром тривалого роздавлювання (краш-синдром).
• Краш-синдром - це патологічний процес, який розвивається в потерпілих у результаті тривалого (4-8 г і більше) роздавлювання м’яких тканин кінцівок уламками зруйнованих будинків, споруджень, брилами ґрунту при обвалах у шахтах і ін.
У протіканні краш-синдрому розрізняють 3-и періоди: 1) ранній (до 3-х діб) з перевагою явищ шоку; 2) проміжний (з 3-ої до 12-ої доби) з перевагою гострої ниркової недостатності; 3) пізній (з 8-12-ої доби до 1-2 міс.) - період видужання, або період, який характеризується перевагою місцевих симптомів.
У розвитку краш-синдрому велике значення мають 3-и патогенетичних фактори: а) больове подразнення; б) травматична токсемія, обумовлена всмоктуванням токсичних продуктів аутолізу тканин з вогнища ушкодження; в) плазмо- і крововтрата, пов’язані із набряком і крововиливами в зоні роздавлених чи довгостроково ішемізованних тканин.
Пусковим механізмом є припинення у перетиснених тканинах кровообігу та відтоку лімфи. Внаслідок цого швидко виснажуються можливості тканинного дихання, виникають аноксія, ацидоз, як наслідок – накопичення проміжних продуктів обміну та вазоактивних речовин, що розширюють судинне русло та збільшують капілярну проникність. Надалі виникають плазмовтрата в прилеглі тканини, руйнування еритроцитів, аутоліз м’язової тканини. Саме в цей період візначається поява у потерпілого агресії, яка зумовлені відчуттям жаху, приреченості, болем, виснаженням стрес-потенціалу та антистресових механізмів. Після звільнення кінцівки кров із магістральних судин починає заповнювати різко розширене судинне русло пошкодженої кінцівки, та, оскільки стінка судини пошкоджена, відбувається проникнення крові зв її межі. Виникають гіповолемія та подальша типова реакція централізації кровообігу. В ушкодженій ділянці настає некроз м’язів, приєднуються інфекційні ускладнення, що формує первинний ендотоксикоз. В кровообіг надходять міоглобін, гемоглобін, калій, креатинін, фосфор, гістамін, т.з. «ішемічний токсин», молекули середньої маси, ейкозаноїди, цитокіни, вільні радикали та ін. Останнім притаманна пошкоджувальна дія та ініціалізація цілого каскаду патологічних змін, зокрема, гострої ниркової недостатності, тромбогеморагічного синдрому, респіраторного дистрес-синдрому, виникають дистрофічні та некробіотичні зміни в печінці, токсичний міокардит, токсико-бактеріальний шок, загострення хронічних захворювань.
У клінічному перебігу виділяють наступні періоди: період компресії; ранній післякомпресійний період (1-3 доби); проміжний період (4-18 доби); пізній період (після 18-ї доби);
Проміжний період характеризується проявами поліорганної недостатності, які зумовлюють тяжкість перебігу гострої ниркової недостатності, прогресуванням гнійно-некротичних процесів у м’яких тканинах та ступенем ендогенної інтоксикації. Розвиваються анемія, вторинні пневмонії, плеврити, ателектази, міокардіодистрофія, міокардити, токсичний гепатит, інтоксикаційний парез кишок, тромбогеморагічний синдром та ін.
У пізньому періоді переважають місцеві симптоми над загальними. Функції нирок поступово відновлюються, нормалізується водно-електролітний баланс, повністю зникає набряк ушкодженої кінцівки. У зоні компресії виникає атрофія м’язів, контрактури, ішемічні неврити, які супроводжуються вираженим больовим синдромом.
Травматичний шок
Розвивається при масивних ушкодженнях м'яких тканин, переломах кіток, забоях, розривах внутрішніх органів. Провідними в розвитку шоку є біль, дія токсичних продуктів розпаду тканин і біологічно активних речовин – гістаміну, калікреїн-кінінової системи. Важливе значення у виникненні шоку має крововтрата, особливо її інтенсивність. Так, при сповільненій крововтраті і зменшенні ОЦК на 20-30 % лише знижується AT, тоді як швидка та інтенсивна крововтрата та швидке зниження ОЦК на ЗО % може призвести до розвитку шоку або смерті. Зменшення ОЦК (гіповолемія) є основним патогенетичним фактором травматичного шоку. Стан кровообігу в організмі контролюють за частотою пульсу і величиною артеріального тиску. Між ними існує прямий зв'язок: з розвитком шоку систолічний тиск знижується, а частота пульсу наростає.
Розрізняють еректильну і торпідну фази шоку. Еректильна фаза дуже коротка, виникає після травми і характеризується напруженням симпатико-адреналової системи. Шкірні покриви бліді, пульс частий, артеріальний тиск підвищений, хворий збуджений.
При торпідній фазі шоку спостерігають загальну заторможеність, низький артеріальний тиск, ниткоподібний пульс. За тяжкістю клінічних проявів торпідної фази шоку розрізняють 4 ступені:
– При шоці І ступеня свідомість збережена, хворий контактний. Артеріальний систолічний тиск знижений до 90 мм рт. ст., пульс помащений. Шкірні покриви бліді, інколи появляється м'язове тремтіння.
– При шоці II ступеня хворий загальмований. Шкірні покриви бліді, вкриті холодним потом. Артеріальний систолічний тиск знижується до 90-70 мм рт. ст. Пульс слабкого наповнення і напруження – 110-120 на хвилину. Дихання поверхневе.
– При шоці III ступеня стан хворого дуже тяжкий: він адинамічний, загальмований, на запитання відповідає сповільнено, не реагує на біль. Шкірні покриви бліді, холодні з синюшним відтінком. Дихання поверхневе, часте. Артеріальний систолічний тиск 70-50 мм рт. ст. Пульс 130-140 на хвилину. В цій стадії припиняється виділення сечі.
– При шоці IV ступеня (передагональний стан) шкіра і слизові оболонки бліді з синюшним відтінком, дихання часте, поверхневе, пульс – ниткоподібний, артеріальний систолічний тиск – 50 мм рт. ст. і нижче.
Тяжкий травматичний шок часто супроводжується порушенням дихання, нудотою, блюванням.
14.Загальна і місцева дія термічних факторів на організм. Патологічні і пристосувально-компенсаторні зміни в патогенезі гіпо- та гіпертермії. Особливості підтримки температурного гомеостазу у дитячому віці.
1. Дія механічних чинників. Поняття про травму.
Поняття про травму вживають в широкому та вузькому розумінні. В широкому розумінні травмою називають будь-яке пошкодження організму і навіть дія на психіку, а у вузькому розумінні - порушення цілісності органу і тканин.
Механічна травма може бути викликана тупим предметом (ушиб, удар), гострим предметом (різані, колоті рани), вогнестрільною зброєю. В результаті механічної травми проходить змертвіння тканин, розмізчення їх, струс, розтягнення перелом кісток. Така травма супроводжується порушенням цілісності кровоносних судин і кровотечею. Наслідки механічної травми різні, вони залежать від об'єму пошкодження, життєвої важливості органу і розміру крововтрат. Одним з найтяжчих наслідків механічної травми є травматичний шок.
При травмі можливі тяжкі загальні зміни і без грубих пошкоджень тканин (струс мозку). Він супроводжується втратою свідомості, ослабленням роботи серця.
Механічна травма порожнин (черевної, грудної, черепної, суглобів) викликає закриті і відкриті пошкодження. Закриті - без дотику з зовнішньою раною. Відкриті рани інфікуються мікробами і сприяють виникненню газової гангрени з прогресуючим змертвінням.
При пораненнях спинного мозку розвивається параліч, утворюються пролежні, порушується виділення сечі і калу.
2. Фізичні чинники. Дія тепла і холоду.
Висока температура навколишнього середовища і окремих предметів викликає місцеву і загальну дію на організм.
Місцева дія виражається опіками, які виникають внаслідок дії гарячих рідин, пару, вогню і т.д. Розрізняють 4 ступені опіку:
1. почервоніння з послідуючим запаленням
2. утворення пухирців
3. змертвіння тканин
4. обвуглення тканин
Ступінь і характер розвитку опіку залежить від способу тривалості і місця дії подразника. Температура 50° діє патологічно. При температурі зовнішнього середовища вище температури тіла тварини виникає перегрівання - гіпертермія. Виникає при перегонах тварин в жару, перевозах, в скупчених приміщеннях.
Гіпертермія протікає в 3 стадії:
1. Захисно-компенсаторна (стадія збудження). Температура в
нормі, це досягається за рахунок рефлекторного зниження теплоутворення і підвищення тепловіддачі. Це виникає в результаті розширення периферичних судин, прискорення дихання, серцебиття, посилене потовиділення, тварина
збуджена, хоче знайти прохолодне місце. Якщо температура діє, то вона підвищується на 1 -2'.
2. Стадія власне хвороби. Дихання і пульс прискорені, периферичні судини розширені, але ці механізми не можуть підтримувати температуру в границях фізіологічної норми. Стан тварини пригнічений.
3. Тепловий удар (шок). Відрізняється від сонячного удару підвищенням температури.
Холод діє місцево і загально. Місцева дія викликає рефлекторний спазм периферичних судин. Це приводить до порушення кровозабезпечення певної ділянки тканин і наступає обмороження. Є 3 ступеня обмороження: 1.блідість шкіри; 2.утворення пухирців; З.змертвіння тканин. Частіше обморожуються периферичні ділянки шкіри (кінцівки, вуха, припуцій).
Загальна дія холоду викликає переохолодження - гіпотермію. Виникнення гіпотермії залежить не тільки від самої причини, але і від стану організму: крововтрати, голодування, ураження теплових центрів. Поява
гіпотермії пов'язана з віком (молоді переносять гірше) і видом (свині замерзають швидше ніж ВРХ, тому, що у ВРХ високе теплоутворення. Найбільш стійка до переохолодження птиця). Протікає гіпотермія в 2 стадії:
1. Захисно-компенсаторна, коли температура тіла в нормі за рахунок посиленого теплоутворення (активізація обміну речовин, робота м'язів, дрижання) й зниження тепловіддачі (звуження периферичних судин, з'єрошення). У людей -гусина шкіра. Підвищений обмін речовин при охолодженні потребує більшої витрати їжі на зігрівання тіла і меншого -на продуктивність, що економічно не вигідно при утриманні тварин в холодних приміщеннях.
2. Стадія охолодження (декомпенсація). В умовах тривалої дії холоду захисні і компенсаторні процеси в організмі виснажуються, температура тіла знижується і наступає смерть. Порушення дихання і кровообігу приводить до кисневого голодування, пригнічення нервової системи. Ці зміни можна назвати охоронними, тому що знижується чутливість клітин мозку до нестачі кисню, інфекції, інтоксикації, дії електроструму і т.д. Охолодження є причиною простудних захворювань.
Дія електричного струму.
Електрострум - причина відрізняється від інших причин:
1. струм діє при безпосередній дії і через різні предмети;
2. струм може викликати зміни при контакті з тканинами, і на шляху його проходження.
На тварин струм діє загально і місцево. Місцево стум проявляє електромеханічну, електротермічну, електрохімічну дію. Електромеханічна дія струму проявляється утворенням газу і пару, які порушують цілісність тканин (рани, переломи). Ці зміни проявляються при дії блискавки.
Електротермічна дія пов'язана з утворенням тепла при проходженні через тканини (особливо кісткової). Проходить розплавлення кісток з послідуючим застиганням.
Електрохімічна дія пов'язана з електролізом в тканинах, порушенням в них хімічних процесів.
Загальна дія струму на організм проявляється в дві фази:
1. короткочасне збудження, яке характеризується прискоренням дихання, пульсу, підвищенням кров'яного тиску:
2. пригнічення, зниження тиску, сповільнення дихання і серцебиття.
Смерть від електротравми викликається паралічем серця чи дихального центру.
Дія електроструму на організм залежить:
1. від характеру електроструму тобто його сили, напруги, характеру (постійний чи змінний) частоти;
2. від стійкості організму, від виду тварини (коні чутливіші від ВРХ), від віку тварини (молоді переносять слабше), від стану (вгодованість, індивідуальні особливості), від стану ц.н.с. (збудження посилює дію струму, наркоз і сон - знижують):
3. від тривалості дії електроструму;
4. від шляхів проходження електроструму в організмі;
5. від
стану шкірного покриву.
Дія променевої енергії.
У виникненні патологічного процесу визначну роль відіграють ультрафіолетові та інфрачервоні промені. Ультрафіолетові проявляють хімічну дію на організм. В великих дозах вони порушують обмін речовин (особливо білковий), посилюють процеси розпаду в тканинах, посилюють чутливість до світла (у ВРХ, овець, свиней, коней при поїданні конюшини і гречки в період цвітіння). При поїданні цих рослин в організмі утворюються рослини (фотодинамічні), які підвищують чутливість тварин до світла. Інфрачервоні промені проявляють теплову дію викликаючи опіки, тепловий чи сонячний удар.
Дія атмосферного тиску.
Зниження атмосферного тиску відчувають при підйомі вгору (гірська хвороба). Патологічні зміни, які виникають при цьому, обумовлені трьома факторами: зменшенням парціального тиску кисню у вдихуваному повітрі, зниженням атмосферного тиску і нестачею вуглекислоти в організмі тварин при прискореному диханні.
При зниженні атмосферного тиску, гази, які знаходяться в організмі, розширюються. Точка кипіння крові і інших біологічних рідин організму знижується, що вони можуть закипати при температурі тіла тварини. Повітря розширюється в кишечнику, викликаючи здуття (метеоризм). Відмічається
кровотеча з носу внаслідок розриву дрібних судин. Нестача вуглекислоти, викликана прискоренням дихання, приводить до зміни РН крові в лужну сторону. Порушується функція ферментних судин і обміну речовин. Виникає вона у непристосованих до гір тварин, і характеризується пригніченням, розширенням кровоносних судин шкіри, слизових рота, носа, вух, прискоренням дихання, серцебиттям, збільшенням в крові еритроцитів. Підвищення атмосферного тиску (на 2-4 атм.) проводиться експериментально. Швидке переміщення тварин з гір в нормальні фізіологічні умови, супроводиться розчиненням в крові газів (азот, кисень). Вони не встигають вийти з крові, нагромаджуються і викликають газову емболію.
Дія іонізуючого променя.
Основні джерела іонізуючого випромінювання - рентгенівське і радіоактивне. Енергія іонізуючого променя перевищує енергію взаємозв'язку молекул і атомів. Потрапляючи в організм, промені розривають слабкі зв'язки молекул (вода Н20 розпадається на іони Н+ і О" ). Ці іони вступають у взаємозв'язок з іншими молекулами, змінюючи їх природу, структуру. Іони води і інших молекул діють на ДНК, змінюють ферментативні реакції синтезу і розпаду молекул, порушуючи поділ клітин. Найбільш чутлива кровотворна тканина епітелій травних і статевих залоз, ендотелій судин, хрящова, кісткова і нервова тканини. Нервові клітини не діляться і тому гинуть останніми. В
першу чергу з'являється нестача в крові лімфоцитів та інших клітин, знижуються захисні реакції: фагоцитоз, утворення антитіл. Це сприяє активізації умовнопатогенної мікрофлори. Із-за зниження кількості тромбоцитів і ураження ендотелію судин - крововиливи. Це все характеризує променеву хворобу. Вона протікає гостро і хронічно. Смерть наступає під час опромінення, або через кілька днів після опромінення. При хронічному перебігу анемія, зниженням лейкоцитів, крововиливи, пневмонія, блювота, пронос, запалення язика, ротової порожнини, що викликає порушення сприймання корму.
3. Дія хімічних чинників.
Хімічні речовини в залежності від дози, являються причиною різних отруєнь. До отруйних речовин належать: кислоти, луги, солі важких металів (ртуті, свинця, міді, олова) , алкоголь, ефір, хлороформ, фенол, ціаністі сполуки, рослинні отрути (алкалоїди, глюкозиди), тваринні отрути (змій і комах), продукти гниття і бродіння.
Більшість отрут вибірково діють на органи і тканини. Миш'як, фосфор, солі важких металів переважно впливають на органи травлення, стрихнін і миш'як на нервову систему, аперетянка - на серце, чадний газ на кров. Розрізняють місцеву і загальну дію отрут. Місцева дія проявляється запаленням і змертвінням. Загальна - в розладах всіх систем організму, особливо нервової. Дія хімічних речовин залежить від характеру отрут, дози, від виду, статі, віку, вгодованості тварин. ВРХ чутлива до солей важких металів, хлороформу; коні - до рослинних отрут; птиці - до солі. Особливістю дії хімічних речовин є звикання до них і нагромадження в організмі при повторних введеннях. Звикання сприяє зменшенню проникнення слизових, а
значить і всмоктування отрут. При цьому швидко руйнуються хімічні речовини в організмі, утворюються анти отрути, і отрути виводяться.
4. Дія біологічних чинників.
До них відносяться патогенні мікроби, віруси, гельмінти, комахи, найпростіші, гриби. Вони викликають інфекційні та інвазійні хвороби. Можливість захворювання тварин залежить від вірулентності мікробів, стійкості тварин продуктам своєї життєдіяльності.
Інфекційні хвороби передаються при безпосередньому контакті від хворої до здорової тварини через повітря, корм, воду, різні предмети, обслуговуючим персоналом, комахами, гризунами.
Інвазійні хвороби викликаються паразитичними червами, найпростішими, комахами.
Температурний гомеостаз у дітей залежить від вікових особливостей та ступеня зрілості усієї системи терморегуляції. Остання визначається стабільністю ректальної температури при температурі повітря 20,0- 22,0°С, наявністю різниці між ректальною та аксілярною температурою, добовим ритмом температури тіла, розвитком лихоманки при інфекційних захворюваннях . Ця залежність особливо чітко проявляється в неонатальному періоді. Необхідна для підтримання температури тіла кількість тепла продукується у доношеної дитини зразу після народження . Теплопродукція у новонародженого складає 1.5 ккал на 1 кг маси тіла за 1 годину. Посилена продукція тепла відбувається шляхом окислення бурої жирової тканини під впливом холодового подразнення дитини, а також за рахунок впливу гормонів щитоподібної залози, наднирників та інтерлейкінів, чинника некрозу пухлин і інтерферону, ліпополісахаридів (бактерійного походження) – так званих ендо- і екзопірогенів.
На молекулярному рівні це відбувається таким чином. Підвищення теплопродукції після народження забезпечується активацією окислення в мітохондріях жирових клітин вільних жирних кислот, рівень яких зростає при підвищенні тонусу симпатичної нервової системи, стимуляції катехоламінами адренорецепторів і активації протеінкінази А, яка підвищує активність фермента ліпази бурої жирової тканини. Більш сильне та тривале підвищення теплопродукції досягається дією гормонів щитоподібної залози на мітохондріальні процеси окислення в жирових клітинах. При цьому посилення теплопродукції досягається прискоренням базових метаболічних процесів (основного обміну) і активацією механізмів факультативного термогенезу, що забезпечують посилення теплоутворення в умовах пониження температури середовища. Від рівнів тиреоїдних гормонів залежать інтенсивність базисного метаболізму в організмі та термогенна функція бурої жирової тканини. Окислення жирних кислот бурої жирової тканини, маса якої у доношеного новорожденого становить близько 2 % від маси тіла (25–35 г), здійснюється без значного синтезу макроергів та з максимально можливим утворенням первинного тепла.
Біла жирова тканина новонарожденого також здатна до прямого теплоутворення, але в значно меншому ступені. При допомозі механізму нескорочуваного термогенезу рівень теплопродукції може бути збільшений в декілька разів у порівнянні з рівнем основного обміну. Разом з тим у доношених дітей запаси теплоутворюючої жирової тканини, в тому числі бурої, швидко зменшуються, досягаючи мінімуму до 3–4-го тижня після народження . Чим вищі сироваткові рівні Т4 і Т3, тим вищий рівень експресії генів в ядрах адіпоцитів бурої жирової тканини, що відповідають за синтез білка термогеніну, який розділяє процеси дихання та фосфорелювання та понижує синтез АТФ в мітохондріях і збільшує теплоутворення [6, 9, 15, 16]. Т3 впливає на термогенез в бурій жировій тканині через модуляцію активності фермента дейодинази Д2, від якого залежать швидкість утворення з Т4 інших активних форм тіреоїдних гормонів та їх метаболічного розщеплення в тканинах. Максимальна стимуляція експресії генів термогеніну досягається одночасною дією тиреоїдних гормонів і катехоламінів. На момент народження ця дія досягає своєї найбільшої вираженості та забезпечує умови для максимальної термогенної активності бурої жирової тканини в ранньому постнатальному періоді. Про дозрівання центральних гіпоталамічних механізмів терморегуляції у дітей може вказувати встановлення правильного добового ритму температури тіла, що відбувається до 1,5-2-місячного віку. Гіпоксія, внутрішньочерепна травма, інфекції, ураження ЦНС та її аномалії можуть бути причиною порушення функції центрального апарату терморегуляції . Плід, що знаходиться в утробі матері при відносно постійній температурі її тіла, не потребує власної терморегуляції. Тепло, що утворює організм плода, передається через плаценту крові матері, і температура крові, що тече від плода до плаценти, на 0,3-0,5°С вища, ніж крові, що тече до плода. Показник теплопродукції плода перед пологами складає 10-15 % від показника теплопродукції матері. Посилена продукція тепла відбувається також за рахунок впливу інтерлейкінів, чинника некрозу пухлин та інтерферону, ліпополісахаридів (бактеріального походження) – так званих ендо- і екзопірогенів. При цьому знижується частота імпульсної активності теплочутливих нейронів, які зменшують або збільшують величину настановчої точки. Окрім того, для новонароджених дітей характерна знижена чутливість центру терморегуляції до лейкоцитарного пірогену, в зв’язку з чим новонароджені діти нездатні перебудувати температурний гомеостаз за типом лихоманки.
Доношені новонароджені вже здатні підтримувати постійну температуру тіла. Однак можливе транзиторне порушення теплового балансу внаслідок, з одного боку, недосконалості процесів терморегуляції (переважання тепловіддачі над теплопродукцією), з іншого – через підвищення або зниження температури навколишнього середовища. Окрім більш високого градієнту температур між поверхнею тіла і зовнішнім середовищем, у дітей є ще ряд анатомо-фізіологічних особливостей, що зумовлюють інтенсивну тепловіддачу.
Незрілість системи терморегуляції найбільш виражена у недоношених немовлят. Для них особливо характерна висока тепловіддача, яка пов’язана з великою поверхнею тіла на одиницю маси, що потенційно збільшує тепловтрати. А також у цих немовлят знижена теплопродукція унаслідок малого вмісту або відсутності бурої жирової клітковини (менше 1 % при 8 % у доношених дітей) . Крім того незріла нервова система та недостатні запаси глікогену у печінці нездатні забезпечити адекватну відповідь на охолодження. При значній мірі недоношеності, коли у новонароджених спостерігається різного ступеня вираженості гіпотиреоз, а маса бурої жирової тканини складає менше 1 % від маси тіла, теплопродукція знижена. Це може сприяти розвитку гіпотермії, якщо не створені умови для обмеження тепловтрат . У дітей, котрі народились недоношеними, в асфіксії або травмованими при народженні, спостерігається значне зниження температури тіла, що може зберігатись декілька діб.
Транзиторна гіпотермія – це зниження температури тіла, обумовлене зміною температури навколишнього середовища дитини. При народженні температура оточуючого середовища нижча, ніж в утробі матері на 12-15°С. Це призводить до того, що в перші 30 хвилин життя температура шкірних покривів кінцівок може знижуватись на 0,3°С в 1 хвилину, а в прямій кишці на 0,1°С, досягаючи мінімальних величин через 30-60 хвилин. У здорових дітей температура тіла незабаром після народження підвищується. До 5-6 години життя встановлюється гомойотермія.
Холодовий стрес – охолодження новонародженої дитини до температури 36,0-36,4°С. Гіпотермія – охолодження новонародженої дитини до температури < 36,0°С. Розрізняють помірну (35,0-36,4°С) і важку (< 35,0°С) гіпотермію. Транзиторна гіпертермія виникає, як правило, на 3-5 день життя, частота її становить 0,3-0,5 %. Температура тіла може підвищуватися до 38,5-39,5°С. Генез транзиторної гіпертермії зі зневодненням . Разом з тим існує точка зору, що найпоширенішими чинниками цього стану є катаболітична спрямованість обміну речовин та гіпернатріємія.
15.Патогенетичне обгрунтування застосування методу терапевтичної гіпотерміі новонароджених при асфіксії.
Перинатальна
асфіксія або гіпоксично-ішемічна енцефалопатія, викликана кисневим голодуванням
при пологах, пов'язана з високим ризиком смерті новонароджених або такими
проявляються пізніше і ведуть до інвалідності неврологічними порушеннями, як
церебральний параліч.
Перинатальна гіпоксично-ішемічна енцефалопатія (ГІЕ) зумовлена недостатнім
надходженням кисню в тканини мозку, що пов’язане як зі зниженням вмісту кисню в
артеріальній крові, так і зі зменшенням мозкового кровообігу . Це
призводить до каскаду патологічних процесів у ішемізованій тканині мозку:
глутамат-кальцієвої ексайтотоксичності, запалення та оксидантного
стресу . Такі несприятливі події в мозку новонародженого впливають на
подальший розвиток дитини і призводять до тривалих тяжких неврологічних
дефектів, таких як розумова недостатність, мовні порушення, труднощі у
навчанні, рухові розлади.
ОСНОВНІ МЕХАНІЗМИ ФОРМУВАННЯ ГІПОКСИЧНО-ІШЕМІЧНОЇ ЕНЦЕФАЛОПАТІЇ
Серед перинатальних чинників ризику розвитку ГІЕ найбільше значення мають гіпоксія та ішемія головного мозку як наслідок системної гіпоксемії і зниження мозкового кровообігу, внаслідок чого виникають структурні ушкодження головного мозку. Зменшення мозкової перфузії спричиняє ураження білої речовини перивентрикулярної ділянки у недоношених дітей і кори головного мозку — у доношених. Гіпоперфузія мозку призводить до лактат-ацидозу та розвитку енергетичного дефіциту. Виділяють декілька фаз енергетичної недостатності при ГІЕ.
Фаза первинної енергетичної недостатності (0—6 год після епізоду ішемії-гіпоксії) відбувається внаслідок зниження мозкового кровотоку, що призводить до зменшення надходження кисню і глюкози до клітин. Через нестачу кисню клітини нервової тканини переходять до анаеробного метаболізму. Це призводить до накопичення молочної кислоти і зменшення кількості АТФ. Порушення клітинного гомеостазу спричиняє накопичення всередині клітини йонів натрію, кальцію, води і збуджувальних нейромедіаторів, що запускає «ексайтотоксично-окисний каскад». Підвищення рівня внутрішньоклітинного кальцію також відбувається в результаті надмірного стимулювання рецепторів нейромедіаторами та деполяризації мембрани. Подальше надходження в клітину йонів кальцію активує ліпази, що призводить до збільшення пулу вільних жирних кислот, підвищеної активації нейрональної синтази оксиду азоту, надлишкової продукції вільних радикалів та дисфункції мітохондрій. Унаслідок мітохондріальної дисфункції запускається апоптоз або некроз клітин. Припускають, що клітини гинуть шляхом апоптозу, якщо енергетичні запаси неповністю вичерпані, а некротична загибель клітин відбувається тоді, коли запас енергії повністю вичерпано .
Фаза вторинної енергетичної недостатності відбувається через 6—48 год після гіпоксично-ішемічного епізоду і також призводить до надмірного вивільнення збуджувальних нейротрансмітерів та вільних радикалів, виснаження запасів АТФ, але відрізняється від первинної фази тим, що не залежить від церебрального ацидозу.
Особливо шкідливим для головного мозку новонароджених є оксидантний стрес через низьку концентрацію антиоксидантів і високе споживання кисню під час переходу від плода до неонатального життя. Новонароджені також мають високі концентрації ненасичених жирних кислот, які розщеплюються з утворенням значної кількості вільних радикалів . Низька здатність головного мозку новонароджених усувати вільні радикали і підвищена чутливість до них призводять до пошкодження нервової тканини .
Протягом третьої фази пошкоджувальні чинники спричиняють подальше ушкодження нервової тканини. Під час цієї фази запускаються механізми запалення та епігенетичні зміни, які призводять до порушення росту аксонів, нейрогенезу та синаптогенезу.
16. Механізм патогенної дії іонізуючого випромінювання на організм. Радіочутливість тканини. Загальна характеристика форм променевого ураження.
Дія іонізуючого випромінювання на організм людини може бути зовнішньою, внутрішньою (якщо радіоактивна речовина потрапила в організм людини при вдиханні чи з їжею) та комбінованою. Ступінь радіаційного ураження залежить від виду випромінювання, тривалості та дози опромінення, фізико-хімічних властивостей радіоактивної речовини та індивідуальних особливостей організму людини.
Патогенез:
Головною ланкою патогенезу радіаційного ураження є ушкодження клітин.
Прямі механізми: безпосередній вплив на макромолекули й надмолекулярні структури клітини. Енергія іоніз. радіації спричиняє іонізацію молекул, розрив найменш міцних зв’язків, утвор. вільних радикалів. Найуразливішими є молекули ДНК.
Варіанти ушкодження:
1) утвор поперечних зшивок між молекулами ДНК і білків, між двома ланцюгами самої ДНК;
2) окиснення і розщепл. азотистих основ;
3) руйнування зв’язків між вуглевод. компонентом і фосфорною к-тою;
4) розрив одного/обох ланцюгів молекули ДНК (розрив хромосом).
Непрямі: при проходж. струменів через клітину відбув. іонізація молекул і атомів. Молекули води іоніз. – радіоліз води – джерело вільних радикалів водню і гідроксилу.
Радіочутливість - це здатність живих організмів реагувати у відповідь на подразнення, викликане поглинутою енергією іонізуючого випромінення. Це його здатність реагувати на мінімальні дози.
Радіочутливість тканини прямо пропорційна її мітотичній активності та обернено пропорційна ступеню диференціювання клітин, з яких вона утворена. Отож, тканини, що діляться, є дуже радіочутливими, а більш диференційовані є більш резистентними.
Так у ссавців печінка, м’язи, мозок, кістки, хрящі та сполучна тканина відносяться до резистентних, оскільки ці тканини у дорослих проявляють низьку проліферативну активність і складаються із спеціалізованих зрілих клітин.
Навпаки ж клітини кісткового мозку, гермінативні клітини яєчників та сім’яників, епітелій кішківника та шкіри є сильно радіочутливими. Виключення: овоцити та лімфоцити хоч і не діляться, але є радіочутливими.
Променеве ураження — ушкодження органа, тканини або системи органів, спричинене дією іонізуючого випромінювання і деякими іншими видами випромінювання, наприклад — інфрачервоного, ультрафіолетового тощо. Зумовлене найчастіше біологічною дією іонізуючого випромінювання. Ушкодження, що виникають під час променевого ураження іонізуючим випромінюванням, породжують променеву хворобу. Променеве ураження від інфрачервоного випромінювання проявляється тепловими опіками, перегріванням. Ультрафіолетове випромінювання виявляє головним чином хімічну дію.
Ураження може бути місцевим (промен. опіки, промен. катаракта) і загальним (промен. хвороба). Від перебігу: гострим/хронічним. За часом настання: раннім/віддаленим. За ймовірністю розвитку: нестохастичні(залежні від величини поглинутої дози)/стохастичні(незалежні).
17. Місцева і загальна дія на організм іонізуючого випромінювання. Гостра променева хвороба, її форми. Патогенез кістково-мозкової форми гострої променевої хвороби. Віддалені наслідки дії іонізуючого випромінювання.
Ураження може бути місцевим (промен. опіки, промен. катаракта) і загальним (промен. хвороба).
Гостра променева хвороба розвивається при короткочасному (кілька хв.- 1-3 дні) опроміненні всього тіла в дозі, що перевищ. 0,5 Гр (50 рад).
Причини: 1) застосув. ядерної зброї; 2) техногенні аварії (атомні електростанції).
Форми: 1) Кістково-мозкова (0,5-10 Гр ); 2) Кишкова (10-50 Гр ); 3) Мозкова (50-200 Гр). Понад 200 Гр – миттєва смерть.
Кістково-мозкова форма:
4 періоди:
1) Період первинних реакцій, з боку
- ЦНС (збудження, запаморочення, гол. біль);
- с-ми кровообігу ( зміни АТ, почервоніння шкіри);
- с-ми травлення ( нудота, закрепи);
- перифер. крові ( лімфопенія).
2) Латентний , період уявного благополуччя ( репаративні процеси, відносно добре самопочуття, лейкопенія, зменш. к-сті гранулоцитів);
3) Період виражених клін. ознак (гематолог. синдром, інфекц. ускладнення, гемораг. синдром, гіпо-/апластична анемія, астенічний синдром, кишковий синдром);
4) Період завершення хвороби (одужання/смерть).
Головною ланкою патогенезу кістк-мозк. форми є ураження кровотворних клітин і лімфоїдної тканини, кишк. – епітел. клітин травного тракту, мозк. – нейронів головного мозку.
Віддаленими наслідками радіаційного ураження вважають:
Мутації в статевих і соматичних клітинах. В статевих – розвиток спадкових хвороб (генетичні дефекти – найвіддаленіші наслідки); в соматичних - злоякісні новоутворення - лейкози, ракові пухлини, променеву катаракту, нефросклероз - хвороба, що виникає внаслідок морфологічного переродження тканин і судин нирок при ураженні їх радіоактивними речовинами при їх виведенні з організму, а також скорочення тривалості життя і прискорення старіння, що реалізується в останні періоди життя.
18. Патогенний вплив надмірної та недостатньої інсоляції ультрафіолетовими променями. Фотосенсибілізація.
Ультрафіолетове випромінювання має теплову, фотохімічну і слабку іонізуючу дію.
Надмірна інсоляція УФП:
При місцевій його дії може розвиватися еритема (почервоніння). Спочатку вона короткочасна, з'являється через кілька хвилин і швидко проходить. Виникає рефлекторно і пов'язана з тепловим впливом ультрафіолетового випромінювання (первинна еритема). Через кілька годин з'являється стійке почервоніння з явищами набряку, болем, загальними змінами (слабкість, головний біль, інтоксикація). Це вторинна еритема. Вона обумовлена утворенням і вивільненням у тканину біологічно активних речовин (гістаміну, серотоніну, кінінів, простагландинів), а також утворенням токсичних продуктів при розпаді тканинних білків, викликаному ультрафіолетовим випромінюванням.
Фотохімічний ефект – пігментація – сонячний загар.
Передчасне старіння шкіри (зморщування, втрата еластичності, нерівномірна пігментація).
Тривалий вплив ультрафіолетового випромінювання може мати генетичні наслідки: мутації, розвиток злоякісних пухлин шкіри й поверхневих тканин.
Недостатня інсоляція: «світлове голодування».
Фотосенсибілізація — це підвищення чутливості організму до дії ультрафіолетового випромінювання. Розрізняють екзогенні (еозин, риванол, акридин, хінін, сульфаніламіди) і ендогенні фотосенсибілізатори (жовчні кислоти, гематопорфірини, холестерол, білірубін). Вважають, що під впливом фотосенсибілізаторів ультрафіолетові промені починають взаємодіяти з тими молекулами тканин, з якими під час відсутності фотосенсибілізаторів вони не взаємодіють.
19. Дія на організм високого та низького атмосферного тиску. Патогенез синдромів компресії і декомпресії. Вибухова декомпресія.
Впливу зниженого атмосферного тиску людина зазнає під час піднімання на висоту: у літаку, у горах. На організм людини за цих умов діють такі патогенні чинники:
1. Власне зменшення атмосферного тиску. Цей фактор зумовлює розвиток синдрому декомпресії, що виявляє себе болем у вухах і лобових пазухах у результаті розширення повітря, яке заповнює їхні порожнини; метеоризмом; кровотечами з носа внаслідок розриву дрібних судин;
2. Зменшення парціального тиску кисню у вдихуваному повітрі є причиною розвитку кисневого голодування (гіпоксії);
Впливу підвищеного атмосферного тиску людина зазнає при зануренні на велику глибину у воду під час водолазних і кесонних робіт. При цьому на організм людини діють такі патогенні фактори:
1. Власне підвищення атмосферного тиску (компресія). Цей фактор викликає вдавлення барабанних перетинок, у результаті чого може з'являтися біль у вухах. При різкому й дуже швидкому підвищенні атмосферного тиску можливий розрив легеневих альвеол. В умовах компресії в крові й тканинах організму розчиняється додаткова кількість газів (сатурація).
2. Азот чинить патогенну дію при диханні стисненим повітрям. При цьому виникають порушення діяльності ЦНС: спочатку легке збудження, що нагадує ейфорію ("глибинний екстаз"), далі - явища наркозу й інтоксикації. Зазначені порушення пояснюють тим, що в результаті сатурації кількість азоту в організмі зростає в кілька разів, причому найбільше він накопичується в органах, багатих на жирову тканину, у тканинах головного мозку, що містять велику кількість ліпідів. Азот у високих концентраціях справляє наркотичну дію, що нагадує дію закису азоту. Щоб уникнути шкідливої дії азоту, цей газ у дихальній суміші заміняють гелієм. Одержують суміш — геліокс.
3. Кисень при підвищенні атмосферного тиску має токсичну дію. Це пов'язано в першу чергу з тим, що в умовах гіпероксії активуються процеси вільнорадикального окиснення, які є одним з механізмів ушкодження клітин. Крім того, як вважають, при гіпероксії порушується виведення із тканин вуглекислого газу, що зумовлює своєрідне їх "удушення".
Синдром компресії – при поступовому підвищенні атмосферний тиск викликає вдавлення барабанних перетинок – біль у вухах.
Синдром декомпресії виникає при швидкому поверненні людини в умови нормального атмосферного тиску після водолазних робіт, робіт у кесонах (кесонна хвороба). При цьому розчинені в крові й тканинах гази (азот, кисень) у великій кількості переходять у газоподібний стан, утворюючи величезну кількість бульбашок, - відбувається десатурація. Бульбашки газів, затримуючись у крові й тканинах, можуть закупорювати кровоносні судини, справляючи тиск на клітини, подразнюючи рецептори (газова емболія). Клінічна картина такої хвороби визначається локалізацією газових бульбашок. Найчастіше виникає біль у суглобах, свербіж шкіри; у важких випадках - порушення зору, параліч, втрата свідомості.
Вибухова декомпресія виникає у випадку швидкого перепаду атмосферного тиску від нормального до зниженого, що буває при розгерметизації висотних літальних апаратів (літаків, космічних кораблів). У розвитку цього синдрому має значення баротравма легень, серця й великих судин внаслідок різкого підвищення внутрішньо-легеневого тиску. Розрив альвеол і судин сприяє проникненню газових бульбашок у кровоносну систему (газова емболія). У крайніх випадках настає миттєва смерть унаслідок закипання крові й інших рідин організму, а також у результаті блискавичної форми гіпоксії.
20. Хімічні патогенні чинники як проблема екології і медицини. Токсичність, канцерогенність, тератогенність хімічних сполук. Екзоінтоксикації. Патофізіологічні аспекти паління, алкоголізму і наркоманії. Значення шкідливих звичок у дитячому віці.
За походженням розрізняють екзогенні токсичні речовини природного походження та синтетичні. До природних відносяться токсиканти біологічного походження (бактеріальні токсини, отрути рослинного та тваринного походження), неорганічні сполуки та органічні сполуки небіологічного походження. Серед екзогенних токсичних речовин виділяють окрему групу ксенобіотиків – штучно синтезованих речовин, які не зустрічаються у природі.
Токсичність речовини визначається низкою факторів, а саме: фізичними і хімічними властивостями токсичної речовини; дозою; концентрацією; шляхом та швидкістю проникнення токсичної речовини в організм; віком, статтю, масою тіла, реактивністю організму; харчовим режимом; наявністю захворювань.
Відповідь організму на токсичну дію хімічної речовини характеризується метаболічними, функціональними, морфологічними та клінічними проявами (як наслідок ушкодження), які тісно пов’язані з патогенетичними механізмами дії отрути. Токсичність хімічних сполук залежить від наявності реактивних хімічних груп і проявляє себе на рівні організму в цілому, органів та систем, тканинному, клітинному та субклітинному рівнях.
Серед фізичних властивостей хімічних речовин головними елементами, що визначають ступінь ураження організму, вважають стан агрегації, рівень дисперсності, кристалічний поліморфізм, леткість, розчинність, здатність до іонізації.
Токсичність хімічних речовин для дітей вища, ніж для дорослих.
Токсичний вплив хімічних речовин на рівні людської популяції проявляється зростанням захворюваності, смертності, кількості вроджених вад розвитку, зменшенням народжуваності, порушенням демографічних характеристик популяції, зниженням середньої тривалості життя, культурною деградацією.
Токсичні процеси на рівні цілісного організму характеризуються: хворобами хімічної етіології; транзиторними токсичними реакціями (тимчасова втрата дієздатності, що швидко і самостійно проходить, наприклад, подразнення дихальних шляхів, психодислептичний стан); стійкими змінами реактивності організму до дії фізичних, хімічних, біологічних чинників довкілля, а також до психічних і фізичних навантажень (алергія, імуносупресія, швидка втомлюваність); спеціальними токсичними реакціями з тривалим прихованим періодом, що розвиваються лише у частини популяції (канцерогенез, ембріотоксичність, порушення репродуктивної функції).
Ушкоджуюча дія хімічних речовин на окремі органи і системи проявляється органною токсичністю – нейротоксичністю, гепатотоксичністю, гематотоксичністю, нефротоксичністю. Характерні захворювання органів, функціональні реакції (короткотривале падіння артеріального тиску, збільшення частоти дихання, підсилення діурезу), неопластичні процеси.
Цитотоксичність на рівні клітини проявляється оборотними структурно-фунціональними змінами клітини (зміна форми, рухливості), загибеллю клітин (некроз, передчасний апоптоз), мутаціями. Деталізація порушень функціональних і біохімічних процесів за умов дії хімічних чинників можлива тільки на субклітинному рівні.
Найбільш поширеними проявами токсичного процесу є інтоксикації (отруєння): гострі/хронічні.
Канцерогенами називають речовини, які сприяють утворенню та розвитку ракових захворювань.
Тератогенний вплив охоплює всі вади розвитку, спричинені токсичним впливом речовини на зародок (ембріон) або плід. Зараховуються сюди зміни будови, порушення діяльності, затримання розвитку і менша маса плоду, передчасне народження.
Інтоксикація - патологічний стан, що виникає внаслідок дії на організм токсичних (отруйних) речовин ендогенного та екзогенного походження. Розрізняють екзогенні та ендогенні. Екзогенні зазвичай мають назву «отруєння». Перебіг екзогенної інтоксикації визначається в основному динамікою конкретної отрути, що його викликала, дозою і способом її потрапляння в організм, а також функціональним станом останнього.
Хронічний алкоголізм, наркоманія, і токсикоманія відносяться до хвороб патологічної залежності (ХПЗ). Вона виникає при довготривалому зловживанні нейротропними отрутами - психоактивними речовинами (ПАР). Психоактивні речовини - речовини які впливають на психіку людини.
Патологічна залежність від ПАР – це хронічна хвороба мозку, яка являється результатом взаємодії визначених генетичних, біологічних, особистих, соціальних особливостей пацієнта, фактор навколишнього середовища.
Найпоширенішою шкідливою звичкою у дітей є обгризання нігтів. Як наслідок часті бактеріальні захворювання, підвищена чутливість зубів, біль у суглобах (шия та щелепа), інфекційні захворювання, емоційні чинники.
У підлітковому віці діти вже починають балуватися сигаретами, пробують алкоголь та навіть наркотики. Інші шкідливі звички підлітків:
- проблема сучасності - постійне використання смартфонів, від яких деякі діти не відходять. Ці шкідливі звички в школі призводять до падіння успішності.
- поширеною є залежність від батьків, коли підліток не здатний приймати самостійні рішення. Статистика показує, що такі діти більше схильні до прийому алкоголю, наркотиків і куріння.
- ігрова і телевізійна залежність. Іноді дитині віртуальний світ більш привабливий і цікавий, що негативно відбивається на його соціальному житті.
21. Інфекційний процес, загальні закономірності розвитку. Механізми захисту організму від інфекції
Інфекційний процесс – патологічний процесс, що розвивається в макроорганізмі при взаємодії з його з патогенним мікробом.
Патогенез: 1.Проникнення мікробів у мікроорганізм та їх колонізація.2. Поширення мікробів по макроорганізму-дисемінація. 3. Взаємодія мікробів та продуктів їхньої життєдіяльності з клітинами-мішенями та чинниками захисту макроорганізму від інфекції. 4. Виведення мікробів та продуктів їхнього розпаду з організму назовні.
Механізми захисту: 1. Бар*єри 2. Механізми розпізнавання 3. Механізми знищення 4. Засоби утилізації вбитих мікробів та їх виведення з організму
22.Спадкові та вроджені хвороби. Мутації як причина виникнення спадкових хвороб (види, причини, наслідки мутацій). Мутагенні впливи. Порушення репарації ДНК та елімінації мутантних клітин як фактори ризику накопичення мутацій і виникнення захворювань.
Спадкові хвороби -
хвороби які передаються з покоління в покоління і зумовлені порушенням
генетичного апарату статевих клітин. До вроджених хвороб належать хвороби, що
виявляються відразу після народження (можуть бути генетично детермінованими або
не пов’язані з генетичним порушенням). Мутація – стрибкоподібне стійке
порушення генетичного апарату, не пов’язане з поділом клітини чи звичайною
рекомбінацією хромосом. Розрізняють мутацію соматичну, яка не передається, і
генеративну, яка впливає на спадковість потомства. Мутація може бути шкідливою
та корисною. За причиною розрізняють спонтанну та індуковану. Залежно від
розмірів ушкодження генетичного апарату розрізняють генну та хромосомну
мутацію. Біохімічні наслідки мутації залежать від функції тих білків, синтез
яких порушений.Якщо це ферментопатії то порушення навіть одного ферменту може
клінічно виявитися недоумством, хворобами системи крові. Якщо синтез ферменту
повністю відсутній то імунологічними методами білок не буде виявлений. Може
порушуватися швидкість синтезу ферменту, і тоді зменшується його кількість.
Мутагенні впливи : фізичні мутагени, хімічні, біологічні.
23.Характеристика моногенних хвороб за типом успадкування. Молекулярні та біохімічні основи патогенезу моногенних хвороб з класичним типом успадкування: дефекти ферментів, рецепторів, транспортних, структурних білків та білків, що регулюють клітинний поділ.
це хвороби людини, спадкування яких відбувається за законами Менделя. Їхніми причинами є генні мутації. Типи успадкування моногенних хвороб: 1.аутосомно-домінантний, 2.аутосомно-рецесивний, 3. Зчеплений зі статтю.
Патогенез хвороб з А-Д типом успадкування може бути пов'язаний з 1)зменшенням утворення білкового продукту, 2)синтез аномальних субодиниць білкових молекул,3)поява аномального білка з абсолютно новими властивостями.
А-Р тип успадкування: ферментопатіх, хвороби, пов*язані з порушенням синтезу неферментних білків(коагулопатії), транспортних білків, білково-пептидних гормонів
Успадкування, зчеплене зі статтю: 1) зчеплений з Х-хромосомою рецесивний – гемофілія, дальтонізм, атрофія зорових нервів, юнацька глаукома, відсутність сутінкового зору, глюкозо-6-фосфат-дегідрогеназодефіцитна анемія у жінок. 2) зчеплений з Х-хромосомою домінантний – фосфат-діабет3) зчеплений з У-хромосою – стосуються сперматогенезу
24.Патогенез моногенних хвороб з некласичним успадкуванням. Полігенні (мультифакторіальні) хвороби.
Моногенні хвороби з некласичним типом успадкування: синдром ламкої Х-хромосоми, хвороба Лебера, синдром Прадера-Віллі, синдром Ангельмана, гонадальний мозаїцизм.
Полігенний
тип успадкування – коли ознака в організмі кодується не одним, а декількома
парами генів. Передача такої ознаки не підпорядковується законам Менделя.
Наприклад, так передається колір шкіри, величина кров’яного тиску. При
ушкодженні якого-небудь гена може виникати не сама хвороба, а «схильність» до
неї. Чим більша кількість генів пошкоджена, що визначає дану ознаку, тим більша
імовірність виникнення захворювання. Цукровий діабет може успадковуватися по
аутосомно-домінантному, аутосомно-рецесивному і полігенному типу. Схильність до
діабету визначається антигенним складом тканин організму. Підвищуються антитіла
до ss-клітин підшлункової залози → зменшується виділення інсуліну →
підвищується вміст глюкози в крові → розвиток інсулінозалежного цукрового
діабету.
Приклади: заяча губа, вовча паща, вроджені вади
серця, атеросклероз, артеріальна гіпертензія, ЦД 2 типу, подагра.
25.Хромосомні хвороби, їх етіологія, патогенез. Загальна характеристика синдромів Дауна, Клайнфельтера, Шерешевського - Тернера. Роль хромосомних аберацій в етіології і патогенезі пухлин
Хвороби, що виникають внаслідок зміни кількості чи будови хромосом. В основі розвитку лежать геномні і хромосомні мутації.
Механізми
виникнення:
1) порушення розходження хромосом у батьків при
утворенні статевих клітин;
2) порушення розходження хромосом на ранніх
стадіях дроблення зиготи.
Синдром Дауна – трисомія ХХІ хромосоми (всього
47 хромосом). Характеризується відставанням у рості, певними рисами обличчя
(напіввідкритий рот, великий язик), зниженням імунологічної реактивності,
глибокою розумовою відсталістю.
Синдром Клайнфельтера – захворювання, що
характеризується зміною числа статевих хромосом – ХХУ (47 хромосом всього).
Буває ХХХУ, ХХХХУ та ін. Хворіють чоловіки. Характерні ознаки: високий ріст,
недорозвинені вторинні статеві ознаки, знижений чи відсутній сперматогенез,
значна розумова відсталість. В соматичних клітинах виявляється одне чи більше
тілець Барра.
Синдром
Шерешевського – Тернера – характеризується набором хромосом – 45, статевих 1,
тілець Барра нема, стать хворих – жіноча.
26. Етіопатогенез та прояви кістозного фіброзу (cystic fibrosis, муковісцидоз) у дитячому віці
Муковісцидоз є найбільш поширеним летальним спадковим захворюванням у світі. Муковісцидоз (МВ) («mucus — слиз, viscidus — в'язкий», кістозний фіброз) — системне, генетично обумовлене, аутосомно‑ рецесивне захворювання, яке спричиняється мутацією гена трансмембранного регулятора муковісцидозу і характеризується ураженням залоз зовнішньої секреції (переважно дихальної та травної систем), що призводить до фатальних наслідків. Існує кілька форм МВ: легенева (15–20%), кишкова (10%) і змішана (70%) з одночасним ураженням шлунково‑кишкового тракту та органів дихання.с
Прояви кістозного фіброзу:
Кишкова форма починається в ранньому дитячому віці, часто після переходу на штучне вигодовування, через недостатність панкреатичних ферментів. Через порушення процесів травлення швидко розвивається гіпотрофія і дефіцит жиророзчинних вітамінів. У кишечнику переважають гнильні процеси, що супроводжуються скупченням газів, здут‑ тям живота. Дефекації часті, відзначається поліфекалія. Випорожнення з неприємним запахом, світлі, зі значною кількістю жиру.
Легенева форма обумовлена гіперпро‑ дукцією в'язкого секрету в бронхолегеневій системі і приводить до обструктивного синдрому, приєднання вторинної інфекції. Зовнішній вигляд хворого: бліда шкіра із землистим відтінком, акроціаноз, задишка в спокої, бочкоподібна форма грудної клітки, деформація дистальних фаланг за типом «бара‑ банних паличок», обмеження рухової активності, зниження апетиту, відставання у фізичному розвитку. У бронхіальному вмісті переважно виявляються синьогнійна паличка, золотистий стафілокок і гемофільна паличка.
Етіологія:
Причиною характерних патологічних змін в організмі хворого є наявність мутацій в обох алелях гена, локалізованого на довгому плечі хромосоми 7 (7q31). Цей ген контролює синтез трансмембранного регуляторного білка муковісцидозу (CFTR), який функціонує, як регульований циклічним аденозинмонофосфатом хлорний канал на апікальній поверхні епітеліальних клітин.
Патогенез:
Порушення іонного транспорту через апікальну мембрану клітин епітелію спричиняє зневоднення секрету екзокринних залоз.
Порушення підшлункової залози призводить до згущення секрету і закупорки вивідних проток, зменшення кількості бікарбонатів, зниження активності панкретатичної ліпази. Стадія повного рубцювання підшлункової залози може спостерігатися у дітей вже у віці 2-3 років.
Виникнення склерозу у підшлунковій залозі та інших органах у хворих на муковісцидоз обумовлено не тільки застоєм слизового секрету і поганим його відтоком; важливіше значення мають процеси порушення обміну у сполучній тканині, яка формується, порушення функцій фібробластів — основного функціонуючого елементу незрілої сполучної тканини.
Ураження бронхіального дерева супроводжується зміною якісного складу муцину і співвідношення води і макромолекул, збільшення в'язкості слизу епітеліальної оболонки бронхів.
27. Принципи діагностики спадкових хвороб. Цитологічні методи. Методи діагностики ДНК. Принципи профілактики і лікування спадкових хвороб.
Загальні принципи діагностики спадкових хвороб:
1. При обстеженні хворого необхідно застосовувати клініко-генеалогічний метод. 2. Рецидивуючі, хронічні захворювання, що тривалий час погано лікуються, особливо в дитячому віці, часто відносяться до спадкових форм. 3. Наявність у хворого специфічних симптомів, що рідко зустрічаються, або їх поєднання є підставою думати про природжений чи спадковий характер захворювання 4. Патологічні зміни, які охоплюють багато органів та систем, наводять на думку про спадкову (або уроджену) патологію. 5. Про спадкове захворювання можна думати в тих випадках, коли хвороба має уроджений характер. Як зазначено вище, уроджений характер патологічних ознак не завжди свідчить про спадкову природу захворювання, але багато спадкових хвороб є результатом хромосомних аномалій або генних мутацій і формуються вже внутрішньоутробно
Спадкові хвороби — це хвороби, зумовлені порушеннями спадкової інформації (мутаціями), отриманими організмом зі статевими клітинами своїх батьків.
4. Цитологічні методи: дослідження каріотипу в ядрах клітин, що діляться, вивчення статевого хроматину в клітинах слизової оболонки рота, дослідження "барабанних паличок" у ядрах ссгментоядерних нейтрофілів.
Методи дослідження ДНК - різноманітні молекулярно-генетичні методи . Найпоширеніші з них:
■ FISH (флуоресцентна гібридизація in situ) — виявлення генетичних
мутацій за допомогою спеціально помічених фрагментів ДНК-зондів;
■ ПЛР (полімеразна ланцюгова реакція) — дуже точний метод, дозволяє
виявити інфекцію в організмі, навіть якщо її присутність ледь помітна;
■ CGH (порівняльна геномна гібридизація) — застосовується в онкології.
Лікування хворих зі спадковими хворобами та спадковими схильностями:
1. Симптоматичне лікування – терапевтичне (медикаментозне), хірургічне (видалення або заміна уражених ділянок, наприклад, поліпозу прямої кишки, корекція при незростанні верхньої губи, вовчої пащі, при вроджених вадах серця).
2. Патогенетичне лікування – корекція обміну речовин (призначення дієти з низьким вмістом фенілаланіну при фенілкетонурії запобігає розвитку розумової неповноцінності); звільнення від продуктів обміну, які є субстратом патологічної реакції (виведення заліза при таласемії, гемосорбція при родинній гіперхолестеринемії); хірургічне шунтування (накладення анастомозу між воротною та нижньою порожнистою веною при глікотеріозі, введення антигемофільного глобуліну при гемофілії, інсуліну при цукровому діабеті) і т. ін.
3. Етіотропне лікування. Завдяки успіхам молекулярної біології та генної інженерії, про нього можна говорити як про перспективу: можливим буде включення штучно отриманих генів до геному людини; використання спрямованого хімічного мутагенезу, що дозволить індукувати специфічні мутації в чітко визначеному локусі (одержання зворотних мутацій – від патологічного алеля до нормального); виправлення генетичного дефекту трансформацій у еукаріотів.
До профілактичних заходів спадкових хвороб людини належать: медико-генетичні консультації; пренатальна діагностика, диспансеризація.
Профілактика спадкової патології повинна бути спрямована на : 1. Максимальне усунення впливів променевої, теплової енергії, хімічних речовин, вірусів, особливо на людей дітородного віку. 2. Молодим людям не повинні проводитися гемотрансфузії без гострих на те показів. 3. Спиртні напої, нікотин і наркотики до і під час вагітності згубно впливають на потомство, аналогічно як і гіподинамія, стреси, отже необхідним є запровадження здорового способу життя. 4. Суворий токсико-гігієнічний контроль за випуском кожного нового лікарського препарату і призначення медикаментів вагітним жінкам тільки при певних показах. 5. Проведення пренатальної діагностики і у разі виявлення важких спадкових і вроджених дефектів переривання вагітності на ранніх її стадіях. 6. Пропаганда генетичних знань серед населення.
28.Аномалії конституції як фактор ризику виникнення і розвитку хвороб. Класифікації конституціональних типів за Гіппократом, Сіго, Кречмером, Шелдоном, І.П. Павловим, О.О. Богомольцем, М.В.Чорноруцьким.
Конституція - це комплекс морфологічних, функціональних і психічних особливостей організму, досить стійких, що визначають його реактивність і сформовані на спадковій основі під впливом факторів зовнішнього середовища.
Діатез — це своєрідна аномалія конституції, що характеризується ненормальною реакцією організму на фізіологічні й патологічні подразники. Діатез найчастіше
виявляється в дитячому віці, коли ще недостатньо дозріли механізми гомеостазу.
Класифікація Гіппократа. Залежно від особливостей темпераменту людини і її поведінки в суспільстві виділяють сангвініків, холериків, флегматиків І меланхоліків.
Класифікація Сіго. У її основі лежить принцип переважаючого розвитку тієї чи іншої фізіологічної системи. Розрізняють такі типи: дихальний (респіраторний), травний (дигестивний), м'язовий (мускулярний) І мозковий (церебральний)
Класифікація Кречмера. Пов'язує морфологічні особливості людини з особливостями її психіки і з частотою певних психічних захворювань. Виділяють атлетичний, пікнічний і астенічний типи конституції.
Класифікація М. В. Чорноруцького. З погляду основних функцій і обміну речовин людей поділяють на нормостеніків, гіпостеніків і гіперстеніків.
Класифікація О.О. Богомольця. Ґрунтується на особливостях будови й функції сполучної тканини в організмі. Розрізняють фіброзний, ліпоматозний, пастозний і астенічний типи конституції. Для фіброзного типу характерна щільна волокниста сполучна тканина. Для ліпоматозного — інтенсивний розвиток жирової тканини, для пастозного - переважання набрякової, пухкої сполучної тканини, а для астенічного — ніжної, тонкої мезенхіми.
Класифікація І. П. Павлова. Залежно від співвідношення першої і другої сигнальних систем вищої нервової діяльності людини виділяють два типи: художній (переважає перша система) і розумовий (переважає друга).
29.Старіння. Структурні, функціональні та біохімічні прояви старіння. Прогерія. Сучасні теорії старіння.
Старіння — це біологічний руйнівний процес, що неминуче розвивається з віком і веде до обмеження адаптаційних можливостей організму, розвитку вікової патології і збільшення ймовірності смерті. За темпами розвитку виділяють передчасне (прискорене) і ретардоване (уповільнене) старіння. За причинами розвитку старіння може бути фізіологічним і патологічним. Залежно від рівня біологічної організації виділяють старіння молекул, клітин, органів і систем, організму в цілому.
Структурні прояви старіння:
У процесі старіння на клітинному рівні виявляються ознаки ушкодження. Зменшуються розміри ядра, воно зморщується, з'являються ядерні включення невідомого походження. Збільшується об'єм цитоплазми, відбувається їївакуолізація, з'являється багато включень, що складаються з ліпофусцину й гемосидерину. Зменшується кількість мітохондрій, рибосом, загальна площа цистерн ендоплазматичного ретикулуму. У той же час зростає кількість лізосом, зменшується стійкість лізосомпих мембран. На тканинному і органному рівнях характерні явища атрофії — зменшується кількість функціонально активних клітинних елементів паренхіми. Якщо загальна клітинна маса організму людини у віці 25 років становить 47 % маси тіла, то у віці 70 років - тільки 36%. Одночасно збільшується кількість функціонально неактивних компонентів органів і тканин - розвивається їх фізіологічний склероз. На рівні організму в цілому старіння виявляє себе характерним виглядом старої людини: змінюється шкіра, вона стає тонкою, зі зморшками й пігментними плямами; міняється постава; відбувається атрофія скелетних м'язів і жирової тканини; з'являється сивина.
Функціональні прояви старіння.
У процесі старіння зменшується функціональна активність клітин, тканин, органів і систем; зменшуються їх функціональний резерв і надійність. Нарівні клітин виявляють порушення електрофізіологічних процесів (зміни потенціалів дії й спокою, зменшення швидкості поширення збудження та ін.); змен- шення міграційної здатності клітин; порушення ендо- і екзоцитозу, скоротливості, процесів клітинного поділу, міжклітинних взаємодій і рецепції. Характерними є порушення сенсорної, інтегративної, рухової і вегетативної функцій нервової системи. Функція одних ендокринних залоз (статевих, щитоподібної) пригнічується, діяльність інших (аденогіпофіз) - навпаки, збільшується. Істотних змін зазнає функція системи кровообігу: зменшуються хвилинний і ударний об'єми серця, збільшується загальний периферичний опір і артеріальний тиск, розвивається віковий атеросклероз. Зменшуються статичні й динамічні показники легеневої вентиляції, секреторна й рухова активність органів травлення. Порушуються антитоксична функція печінки, видільні функції нирок. На рівні організму в цілому збільшується стомлюваність, зменшується працездатність і адаптаційні можливості організму.
Біохімічні прояви старіння:
При старінні в клітинах з'являються й накопичуються невластиві ліпофусцин, амілоїд. Утворюються аномальні білки, що мають внутрішньомолекулярні "зшивки" змінюється активність багатьох ферментів. У сполучній тканині з'являється "старий" колаген, погано розчинний, із численними внутрішньо- і міжмолекулярними "зшивками". Порушується функціонування геному клітин: розладнується регуляція роботи генів, виникають мутації, страждають процеси транскрипції. Істотних змін зазнає енергетичний обмін: зменшується інтенсивність тканинного дихання і збільшується активність гліколізу, зменшується вміст АТФ і креатинфос-фату в клітинах; пригнічується активність АТФ-аз. У багатьох структурах збільшується інтенсивність пероксидного окиснення ліпідів. У крові зростає вміст холестеролу, ліпопротеїнів дуже низької і низької густини.
Прогерія - це патологічне старіння, що виникає в результаті дії на організм патогенних факторів. Прикладом є синдром Гетчинсона-Гілфорда. Це аутосомно-ре-цесивне спадкове захворювання. Його перші ознаки з'являються дуже рано, уже на першому році життя. Характерна затримка росту, посивіння волосся, облисіння, шкіра набуває старечого вигляду, розвивається катаракта, атеросклероз. Смерть, як правило, настає на 1-2-му десятиріччі життя. 7
Сучасні теорії старіння:
1.Молекулярно-генетичні теорії старіння є найбільш визнаними у сучасній геронтології. Даний вид концепцій вбачає причину старіння у первинних змінах генетичного апарату клітини.
В свою чергу молекулярно-генетичні теорії поділяють на дві групи.
1.1Л. Хейфлік, який встановив, що у нормі соматичні клітини людини мають обмежений мітотичний потенціал та певну тривалість життя. Для свого дослідження вчений брав клітини сполучної тканини (фібробласти), які культивував після перенесення до поживного середовища. Виявлено строго обмежену кількість поділів (ліміт Хейфліка), після чого культура гинула.
1.2.Генно-регуляторна теорія, започаткована В. Райтом. Згідно неї первинні зміни виникають у регуляторних генах – найбільш активних та найменш захищених структурах ДНК. Проте прямих доказів існування вікових змін ДНК не багато. Останнім часом виникнення старіння пов’язують із тими ділянками ДНК, що з віком зменшуються у розмірах.
2.Теорія помилок (Л. Оргел) ґрунтується на припущенні, що основною причиною старіння є накопичення із віком генетичних ушкоджень в результаті мутацій.
3.Інтоксикаційна теорія (Ч. Бухард) розглядає старіння як результат збільшення рівня токсичних речовин в організмі.
30.Поняття про пренатальну патологію. Гамето-, бласто-, ембріо- і фетопатії. Тератогенні фактори. Критичні періоди в пренатальному онтогенезі. Внутрішньоутробні дистрофії, інфекція, гіпоксія. Хвороби і шкідливі звички матері як причинні або фактори ризику патології плода.
Пренатальна патологія –це захворювання, що включають в себе патологічні процеси людського зародка, починаючи з запліднення і закінчуючи народженням дитини. Гаметопатїї. Виникають до запліднення, під час гаметогенезу. Бластопатії. Формуються в перші 15 діб розвитку зародка (бластоцисти). Ембріопатії. Охоплюють порушення, що виникають після диференціювання ембріобласта до закінчення закладки органів (від 16-ї доби до 12-го тижня). Фетопатії - порушення розвитку плода (фетогенезу). Розрізняють патологію раннього фетогенезу, коли відбувається утворення тонких структур і досягається життєздатність плода (від 12-го тижня до 7-го місяця), і порушення пізнього фетогенезу, коли відбувається становлення функцій плода й одночасно - старіння плаценти (від 7-го місяця до пологів). Тератогенні фактори - це фактори зовнішнього середовища, які, впливаючи протягом вагітності, викликають розвиток уроджених вад. Основні групи тератогенних чинників включають лікарські препарати і хімічні речовини, іонізуюче випромінювання, інфекції, метаболічні порушення і шкідливі звички вагітної. Критичний період - це такий проміжок часу в розвитку організму, коли після чергової стадії визначаються подальші шляхи становлення: всього зародка або окремих його зачатків, їхній вступ у новий етап морфогенезу. Розрізняють два основних критичних періоди: 1) період переходу зародка від стадії дроблення до диференціювання на три зародкових листки, що закінчується імплантацією (1-й тиждень); 2) період закладки органів, у тому числі й плаценти, - період плацентації й великого органогенезу (3—8-й тижні). Критичні періоди характеризуються високою метаболічною активністю й підвищеною чутливістю до дії патогенних факторів. Висока уразливість організму в ці періоди пояснює частий розвиток уроджених вад при дії тератогенних факторів.
Внутрішньоутробна дистрофія найчастіше виникає при розвитку різних патологій. Вона може бути викликана і пізнім токсикозом матері, і паталогічна змінами плаценти, які провокують запальний процес, що загрожує інфекційним захворюванням плоду. Неправильне харчування, прийняття алкоголю і куріння також можуть стати причиною виникнення внутрішньоутробної дистрофії. Зростання дитини всередині утроби матері сповільнюється, малюк худне, знижується психомоторне розвиток і реактивність організму. Загрожує дане захворювання дитині тим, що в підсумку може з'явитися непереносимість різних продуктів, порушення збудливості кори головного мозку, зниження вроджених і набутих рефлексів. Страждають всі життєво важливі органи в тілі дитини.
Внутрішньоутробні інфекції (ВУІ) – це інфекційні захворювання, для яких характерне ураження плода збудниками до пологів, або під час проходження через пологові шляхи, викликаючи в окремих органах запальний процес чи ознаки токсемії. За часом інфікування ВУІ можуть бути анте- чи інтранатальними. Обов¢язковою умовою виникнення ВУІ є наявність у матері вогнища інфекції. Найчастіше це цитомегаловіруси, ентеровіруси, віруси герпетичної та респіраторної групи, краснухи, гепатиту В, імунодефіциту людини.
Гіпоксія внутрішньоутробна – це внутрішньоутробний синдром, обумовлений розвитком комплексу змін , які впливають на плід, що виникають внаслідок недостатнього надходження кисню до його тканин і органів. Ризик розвитку гіпоксії у плоду підвищується при таких захворюваннях матері, як анемія, серцево-судинна патологія, захворювання нирок, органів дихання (хронічний бронхіт, бронхіальна астма), цукровий діабет, токсикоз вагітних, багатоплідна вагітність, інфекційні захворювання, що передаються статевим шляхом. Брак кисню в першу чергу негативно впливає на нервову систему, так як вона є найбільш чутливою до нестачі кисню. При дефіциті кисню з 6 по 11 тиждень відбувається затримка дозрівання головного мозку, порушується будова і функціонування судин. Також від негативної дії гіпоксії страждають тканини нирок, серця і кишечника плоду. Навіть незначна нестача кисню може стати причиною розвитку ураження центральної нервової системи. При важких формах гіпоксії плоду відбувається розвиток некрозу і ішемічного ураження різних органів. Після народження дітей, які внутрішньоутробно страждали від нестачі кисню, у них можуть спостерігатися різні неврологічні розлади, затримка психічного розвитку і важкі соматичні відхилення.
Хвороби і шкідливі звички матері : Вживання вагітною алкоголю у великих дозах може привести до розвитку фетального (враження плоду) алкогольного синдрому – вродженого захворювання, що супроводжується чисельними вадами розвитку, який іноді несумісний із життям. Тютюнокуріння великої кількості цигарок під час вагітності призводить до відставання дитини у фізичному розвитку. Вплив токсичних хімічних речовин. Це можуть бути хімічні чинники, які здатні накопичуватися в організмі жінки та проникати через плаценту, фізичні чинники (вібрація, електромагнітне поле комп’ютера), радіаційний вплив. Якість харчування вагітної жінки. Такий вплив обумовлений, в основному, дефіцитом мікроелементів. Найбільш повно вивчений дефіцит цинка, магнія, міді. Важливо, щоб раціон був збалансований за мікроелементами, вітаміном та білками.
Інфекційні захворювання, які передаються від матері до плоду належать вірусні захворювання (краснуха, цитомегаловірус, простий герпес, грип, епідемічний паротит, вітряна віспа). Особливу групу займають інфекції, які передаються статевим шляхом – токсоплазмоз, сифіліс, хламідіоз, уреаплазмоз. В результаті розвиваються різні ускладнення – мимовільний аборт, народжуються недоношені або мертві діти. Такі вірусні інфекції у матері – як краснуха, грип, вірусний гепатит, поліомієліт, порушують тканинний обмін зародка, що розвивається, призупиняють ріст клітин; в результаті виникає дефект відповідного органу і діти народжуються з каліцтвами. Якщо жінка в перші 2-3 міс. вагітності перехворіла на краснуху, у немовляти можуть виникнути катаракта (помутніння кришталика ока), глухота, уроджені вади серця, пошкодження центральної нервової системи (мікроцефалія – череп малого розміру, спинномозкова грижа, гідроцефалія – водянка мозку), ушкодження кісткової тканини (розщеплення твердого піднебіння, арахнодактилія тощо).
31. Роль реактивності в патології. Реактивність і резистентність: визначення, види, механізми. Залежність реактивності від віку, статі, спадковості, стану нервової та ендокринної систем.
Реактивність - властивість організму відповідати змінами життєдіяльності на впливи зовнішнього середовища
Групова реактивність властива певним групам організмів в межах одного виду. За походженням вона може бути як спадковою так і набутою. Виділяють групи за віком, статтю, конституцією, расою, групою крові, резус-фактором. Можна говорити про реактивність людей з хворобою Дауна чи синдромом Марфана, або такими набутими захворюваннями як наркоманія, СНІД, туберкульоз. Набутою також може бути реактивність людей певних професій: шахтарів, програмістів, спортсменів. Можна виділити групи людей з різною реактивністю залежно від місця проживання, типу харчування, пори року чи часу доби. Останнім часом говорять про групову реактивність бідних та багатих людей (високого та низького соціального статусу).
Індивідуальна реактивність зумовлена спадковими і набутими факторами. Вона залежить від тих умов зовнішнього середовища, в яких організм розвивається, – характеру харчування, кліматичного поясу, від вмісту кисню в атмосферному повітрі тощо.
Реактивність залежить від статі. У жіночому організмі реактивність змінюється у зв’язку з менструальним циклом, вагітністю. Жіночий організм більш стійкий проти гіпоксії, крововтрати, радіального прискорення, голодування.
Відома роль віку при визначенні стану реактивності. Ранній дитячий вік характеризується низькою реактивністю. Це визначається незрілістю нервової, ендокринної й імунної систем, недосконалістю зовнішніх і внутрішніх бар’єрів. Найвища реактивність спостерігається в зрілому віці, поступово знижуючись до старості. Люди похилого віку дуже сприйнятливі до інфекції, у них часто розвиваються запальні процеси в легенях, гнійничкові ураження шкіри, слизової оболонки. Причина цього полягає в ослабленні імунних реакцій і зниженні бар’єрних функцій старечого організму.
Індивідуальна реактивність може бути специфічною і неспецифічною.
Специфічна реактивність виражається в здатності утворювати антитіла на антигенні подразнення. Таким вимогам задовольняє імунологічна реактивність. Вона забезпечує несприйнятливість до інфекційних хвороб (імунітет), реакції біологічної несумісності тканин, підвищеної чутливості.
Неспецифічна реактивність виявляється при дії на організм різних факторів зовнішнього середовища. Вона реалізується за допомогою таких механізмів, як зміна функціонального стану нервової, ендокринної систем, біологічних бар’єрів і системи мононуклеарних фагоцитів (СМФ).
Специфічна і неспецифічна реактивність може бути фізіологічною і патологічною.
Резистентність стійкість організму до дії патогенних факторів
32. Особливості реактивності у дитячому віці.
Аналіз результатів імунологічного обстеження в різних вікових групах (1-ша група — діти до 9 років; 2-га — діти від 10 до 14 років та 3-тя група — діти, доросліші за 14 років) показав, що залежно від віку існують вірогідні відмінності або статистичні тенденції змін переважної кількості імунологічних показників, які вивчались, що наочно наведені на рис. 1. За показниками, що не наведені на рис. 1, не було зареєстровано вірогідних розбіжностей.
Порівняння результатів імунологічного обстеження у дітей першої та другої досліджуваних груп дозволило констатувати вірогідні відмінності у концентраціях лімфоцитів (p = 0,032), сироваткового IgA (p = 0,013), IL-4 (p = 0,02), які з віком збільшуються, та TGF-b1, концентрація якого, навпаки, вірогідно зменшується (p = 0,009) (рис. 1).
З дорослішанням у дітей віком понад 14 років (3-тя група) порівняно з групою дітей віком від 10 до 14 років (2-га група) реєструється (рис. 1) подальше зростання відносної концентрації лімфоцитів (p = 0,05), вмісту CD25+ (p = 0,011), що поєднується зі зниженням абсолютної концентрації лейкоцитів (p = 0,041) та зменшенням умісту цитокінів — IL-2 (p = 0,027), IL-4 (p = 0,04), IL-12p70 (p = 0,006), IL-12р40+р70 (p = 0,006) та TGF-b1 (p = 0,00002) у периферичній крові. Також у дітей 3-ї групи у периферичній крові реєструється вірогідно низький уміст лейкоцитів (p = 0,024) та TGF-b1 (p = 0,002), що поєднується з вірогідно високими концентраціями CD25+ (p = 0,052) та відносною кількістю лімфоцитів (p = 0,024) порівняно з результатами у дітей першої групи спостереження (рис. 1).
Для виявлення характеру розподілення та структури результатів імунологічного обстеження у дітей різного віку з дитячих будинків в періоді клінічного благополуччя залежно від відповідності до існуючих стандартів або діапазону тест-системи проведено індивідуальне порівняння всіх отриманих у даному дослідженні результатів у межах середніх значень, вищих за середні та нижчих за середні (табл. 1), що дозволило виділити дітей різного віку, дані імунологічного обстеження яких відрізнялись від середнього рівня концентрації (норма показника за віком).
Згідно з даними табл. 1, уміст лейкоцитів у 98,1 % всіх спостережень зареєстровано в межах норми, а концентрація лімфоцитів у 80,8 % за відносним вмістом та в 13,5 % всіх спостережень за абсолютною кількістю була вищою за нормальну, при цьому більшість високих результатів була зареєстрована у дітей, доросліших за 10 років (2-га та 3-тя група спостереження).
Концентрація сироваткових IgA, IgM та IgG залежно від фактичного результату мала різний характер — нижчу за віковий уміст було зареєстровано у 25,9; 9,3 та 1,9 % відповідно, для всіх спостережень. Максимально часто зміни щодо вмісту сироваткових імуноглобулінів були зареєстровані в другій групі спостереження. Вищий за віковий рівень було зафіксовано тільки для сироваткових імуноглобулінів IgM та IgG (42,6 та 51,9 % всіх спостережень відповідно), питома вага яких превалювала серед дітей 1-ї та 2-ї груп спостереження порівняно з підлітками.
У групах обстежених дітей із дитячих будинків у періоді клінічного благополуччя вміст CD25+-клітин у периферичній крові як за показником абсолютної кількості (51,9 % даних спостережень), так і за показником оптичної щільності (48,1 %) був вищим за представлений лабораторією норматив, і серед цих дітей переважну більшість становили діти, старші за 14 років (75 %).
Вміст цитокінів (IL-2; 4; 10; 12р70; 12р40+р70) у сироватці крові обстежених дітей у переважній більшості випадків не відрізнявся від нормативу показників тест-системи виробника, за винятком вірогідно не значущих відмінностей розподілення фактичного результату вмісту IL-10, IL-12р70 та IL-4 залежно від одиниць вимірювання (оптична щільність).
У 84,9 % усіх результатів було зафіксовано нижчий за середній рівень TGF-b1, представлений виробником тест-системи (57,75–152,25 AU/мл), але який повністю відповідав діапазону визначення даного цитокіну в сироватці крові (норматив тест-системи виробника DRG ELISA, Germany, щодо вмісту TGF-b1 становить 3–600 AU/мл).
Оцінка результатів дослідження мукозальної імунної системи показала, що концентрація протимікробних білків лактоферину та a-дефензинів 1–3 у ротоглотковому секреті у всіх обстежених дітей відповідала нормативу тест-систем виробника, що в 79,7 % випадків поєднувалось зі зниженням умісту SIgA серед усіх обстежених дітей. Максимально часто дефіцит секреторного імуноглобуліну А було зареєстровано в дітей раннього шкільного віку (1-ша група, 90 %), з дорослішанням питома вага даної складової зменшувалась у три рази (до 30 %) у дітей другої групи спостереження та у 4,5 раза (до 20 %) — у підлітків.
За результатами проведеного кореляційного аналізу описаних вище імунологічних показників, вік обстежених дітей мав вірогідні зв’язки з концентрацією лейкоцитів (р = 0,001; r = –0,4387), лімфоцитів (р = 0,009; r = 0,3599) та вмістом цитокінів: IL-10 (р = 0,03; r = –0,258); IL-4 (р = 0,042; r = –0,2923); TGF-b1 (р = 0,003; r = –0,3999). Стать обстежених дітей мала вірогідні кореляції з умістом лейкоцитів (р = 0,05; r = 0,2776), лімфоцитів (р = 0,042; r = –0,2923); рівнем CD25+-клітин (р = 0,042; r = –0,2923); сироватковим IgM (р = 0,021; r = 0,3135) та TGF-b1 (р = 0,018; r = –0,3243).
Показники вмісту лейкоцитів у обстежених дітей корелювали з умістом IL-10 (р = 0,027; r = 0,3589) та лімфоцитів периферичної крові (р = 0,001; r = – 0,4611), які, у свою чергу, мали прямий зв’язок з умістом IL-10 (р = 0,067; r = 0,3004) та концентрацією CD25+-клітин (р = 0,014; r = 0,3398).
Серед цитокінів рівень IL-12p70 (пг/мл) мав прямий кореляційний зв’язок із IL-4 (р = 0,036; r = 0,3009) та зворотній з TGF-b1 (р = 0,05; r = –0,4662). У свою чергу, вміст TGF-b1 мав вірогідні кореляції з IL-4 (р = 0,027; r = –0,5206); IL-2 (р = 0,05; r = 0,5604) та сироватковим IgG (р = 0,021; r = 0,3298). Цитокіни сімейства IL-12р40+р70 були пов’язані тільки з рівнем IgG (р = 0,06; r = 0,3814), а IL-2 корелював із концентрацією IgМ (р = 0,039; r = 0,5776).
У структурі показників мукозальної ланки імунітету концентрація SIgA в ротоглотковому секреті мала зворотній кореляційний зв’язок зі статтю обстежених дітей (р = 0,006; r = –0,3173) та з такими імунологічними показниками, як кількість лейкоцитів (р = 0,018; r = –0,3520), лімфоцитів (р = 0,081; r = 0,2629) та цитокінів: IL-10 (р = 0,068; r = 0,2996) та IL-4 (р = 0,003; r = –0,8320).
Вміст протимікробних білків лактоферину (р = 0,029; r = 0,2732) та a-дефензинів 1–3 (р = 0,032; r = –0,2686) у ротоглотковому секреті мав зворотні кореляційні зв’язки зі станом бактеріоносійства умовно-патогенної флори на слизових верхніх дихальних шляхів; при цьому рівень лактоферину був обернено пов’язаний з умістом сироваткового IgМ (р = 0,05; r = –0,9461).
Таким чином, отримані результати оцінки імунологічних показників у сироватці крові та вмісту протимікробних білків і секреторного імуноглобуліну А у ротоглотковому секреті дітей шкільного віку з дитячих будинків в періоді клінічного благополуччя свідчать про дисбаланс різних ланок імунної відповіді, зниження мукозального імунітету с
33. Роль фізіологічної системи сполучної тканини в резистентності організму до дії патогенних агентів (вчення О.О. Богомольця). Біологічні бар’єри, їх класифікація.
Біологічні бар’єри - це спеціалізовані тканинні структури, що захищають цілий організм або окремі його структури від впливу патогенних чинників навколишнього середовища і забезпечують підтримання гомеостазу. Біологічні бар’єри поділяють на зовнішні та внутрішні.
Зовнішні бар’єри – об’єднують структури, які контактують безпосередньо з зовнішнім середовищем і/або з речовинами, що надходять ззовні. До них належать шкіра, слизові оболонки, дихальні шляхи (відбувається очищення повітря від пилу та різноманітних шкідливих речовин атмосфери), травний тракт (за рахунок бактерицидного впливу шлункового соку і позбавлення поживних речовин антигенних властивостей), печінка (знезаражує токсичні речовини, які надходять з їжею або утворюються в кишківнику), селезінка, лімфатичні вузли, та інші органи, в яких містяться клітини СМФ.
Шкіра і слизові оболонки є першими механічними перешкодами, які захищають організм від проникнення фізичних, хімічних та біологічних патогенних чинників.
Шкіра – механічний бар’єр для проникнення мікроорганізмів, який працює за рахунок продуктів секреторної функції розташованих у ній залоз (потових, сальних), що мають бактерицидні властивості.
Слизові оболонки – механічний бар’єр, бактерицидна дія секретів, лізоцим, кисла реакція, імуноглобулін А, інтерферон.
Шлунково-кишковий тракт – шлунковий сік, бактерицидна здатність якого пов’язана з соляною кислотою, пристінкове травлення, муциновий шар, що покриває епітеліальні клітини, і їхнє постійне відновлення протягом короткого терміну, антагоністична дія нормальної мікрофлори, коротколанцюгові жирні кислоти, комплемент, лізоцим, інтерферон, лактоферин, секрет ентероцитів з ацидофільними гранулами.
Гуморальні фактори – бактерицидні компоненти крові (лізоцим, інтерферон, комплемент, нормальні антитіла, ферменти, простагландини, тромбоцитоактивуючий фактор, еритрин, С-реактивний білок).
Це далеко не повний перелік неспецифічних захисних білків, які забезпечують підтримання гомеостазу організму.
Внутрішні бар’єри
Внутрішні бар’єри регулюють надходження з крові в органи і тканини необхідних енергетичних ресурсів і своєчасний відтік продуктів обміну речовин (очищення, кліренс), що забезпечує постійність складу фізико-хімічних і біологічних властивостей тканинної (позаклітинної) рідини і збереження їх на певному оптимальному рівні. Одночасно вони перешкоджають надходженню з крові в органи і тканини чужорідних і отруйних речовин.
Існування бар’єрів дозволяє зрозуміти, чому деякі отрути, бактерії, віруси, уражають одні органи і не зачіпають інші. Не зовсім зрозумілу спорідненість, “тропність ” окремих тканин до тих чи інших чужорідних речовин, що потрапили у внутрішнє середовище, в тому числі і до фармакологічних препаратів, можна пояснити особливостями будови їх бар’єрних механізмів. В медицині існує поняття про зони найменшого опору, в яких найчастіше виникають вогнища захворювання. В багатьох випадках утворення таких зон пов’язане з порушенням проникливості гістогематичних бар’єрів.
Гістогематичний бар’єр – це спеціалізована структура, що знаходиться між кров’ю і тканинною рідиною, який регулює обмінні процеси між ними, забезпечуючи збереження постійності складу і фізико-хімічних властивостей тканинної рідини, а також який затримує перехід у неї чужорідних речовин із крові.
Так, наприклад, хворі діабетом інколи скаржаться на біль, що відомий під назвою діабетичного невриту. В останні роки було встановлено, що ці болі виникають в результаті порушення гематоневрального, тобто розташованого між кров’ю і нервовими стовбурами, бар’єру. Через високий вміст цукру в крові він стає проникним для деяких болетворних сполук, що є в крові.
До гістогематичних бар’єрів можуть належати всі без винятку бар’єрні утворення між кров’ю і органами. Основним структурним елементом внутрішніх бар’єрів є стінка кровоносних капілярів, яка складається з ендотелію і базальної мембрани. У різних органах ендотелій капілярів має свої морфофізіологічні особливості, які служать основою для специфічності кожного бар’єру, зокрема його вибіркової проникності. Базальна мембрана, натомість, є більш універсальним утвором і складається з колагену та глікозаміногліканів.
Однак, не можна не визнати, що існують спеціалізовані бар’єри, які мають особливе значення для життєдіяльності організму. До них відносять більш детально вивчені гематоенцефалічний бар’єр (між кров’ю і центральною нервовою системою), гематоофтальмічний бар’єр (між кров’ю і водянистою вологою ока), гематотиреоїдний бар’єр (між кров’ю та колоїдом щитоподібної залози), гематолабіринтний бар’єр (між кров’ю і вушною ендолімфою), гематотестикулярний і гематооваріальний бар’єри (між кров’ю і статевими залозами) і т.д. До гістогематичних бар’єрів належать також бар’єри між кров’ю і рідкими середовищами організму (цереброспинальною рідиною, лімфою, плевральною, синовіальною рідиною). Вони отримали назву гематолікворного, гематолімфатичного, гематоплеврального, гематосиновіального бар’єрів. Плацентарний бар’єр (між матір’ю і плодом), хоч і не належить до гістогематичних бар’єрів, здійснює надзвичайно важливу функцію захисту плоду.
Гістогематичні бар’єри ділять на три групи (В.О.Горбань, М.Н.Зайко): ізолюючі (гематоенцефалічний, гематотестикулярний), частково ізолюючі (гематоофтальмічний та гематотиреоїдний) і неізолюючі (гематооваріальний). Результати їх дослідження показують, що гістогематичні бар’єри дуже адаптовані до структури і функції тих органів і тканин, які вони “опікають”.
34. Порушення фагоцитозу: причини, механізми, наслідки.
Порушення фагоцитозу
Розлади фагоцитозу можуть бути: (а) спадково зумовленими і (б) набутими. Залежно від при чин і суті порушень їх можна позділити на три групи.
I. Порушення, пов'язані з особливостями об'єкта фагоцитозу
До таких відносять:
a) розлади, зумовлені чинниками бактерій, що мають антифагоцитарну дію і в такий спосіб захищають себе від впливу захисних механізмів організму (див. главу 11). Так, збудники деяких інфекційних хвороб-мі- мікобактерії туберкульозу, токсоплазми, бру цели, лістерії, збудник лепри, багато видів найпростіших, перебуваючи у фагосомі, вивільняють речовини, що утруднюють або унеможливлюють злиття фагосоми з лізо сомами, тобто формування фаголізосоми.
Макрофаги при цьому перебувають у стані постійної активації, виділяють вміст лізо сом у тканину й цим самим підтримують вогнище хронічного запалення; б) порушення, які виникають при поглинанні ні неорганічних і деяких органічних (напр., холестеролу) речовин, що не метаболізму ються клітинами. Так, фагоцити, захоплю ючи кристали й пилові частки неорганіч них сполук (кварцу, азбесту, цементу, као ліну, кам'яного вугілля та ін.), не можуть їх ані перетравити, ані утилізувати. При цьо му відбувається ушкодження лізосом, крис тали й пилові частки із загиблих макрофа гів знову потрапляють у тканину, де їх по чинають поглинати нові макрофаги. 1 так усе повторюється спочатку. Постійна заги бель макрофагів і виділення ними в ткани ну вмісту лізосом викликає хронічне запа лення й склероз. За таким “сценарієм" від бувається розвиток захворювань легень, ві домих під назвою пневмоконіозів (силікоз, антракоз, азбестоз та ін.).
Аналогічну картину можна спостерігати і в стінці артерій при розвитку атеросклерозу (див. главу 32). Накопичення холестеролу в ма крофагах спочатку веде до перетворення їх у т. зв. "пінисті" клітини, а потім і до загибелі.
Вивільнені при цьому кристали холестеролу знову поглинаються новими макрофагами і т.д.
В артеріальній стінці розвивається хронічне за запалення, яке веде до формування атеросклероз тичної бляшки.
II. Порушення, обумовлені системами, спря женими з фагоцитозом,-порушення опсоні- зації Значне послаблення фагоцитозу може вини кати при розладах опсонізації. Вважають, що цей процес с найуразливішою точкою в пато генезі порушень фагоцитозу. Клінічні прояви таких порушень при розладах опсонізації на стають раніше і є більш вираженими, ніж при зменшенні кількості фагоцитів.
Причинами, що порушують опсонізація 1 в такий спосіб обмежують фагоцитоз, мо жуть бути:
а) набуті і спадково зумовлені імунодефіцит ти, які виявляють себе зменшенням утво рення антитіл - IgG (див. нижче); б) порушення активації системи комплементу- ту (усіма трьома шляхами), що приво- дять до дефіциту Cзь. Крім того, зменшен ня утворення СШа може бути чинником, що послаблюс хемотаксис лейкоцитів; в) дефіцит фібронектину. Pівень цього бл ка в плазмі крові істотно змінюється при важких травмах, опіках, коли в кров потрап де велика кількість тканинних і мікробних BİAKoain, mo wHMarac umAKoi ix Helkrpani- зації і ділення. При цьому відбуваються активаця фіксованих макрофагів (ендоте аіальних клітин Купфера в печінці) значно зростає споживання шми фібронсктину, зв'язаного з такими відходами. Як наслі док, концентрація цього опсоніну в плазмі крові падає.
I. Порушення, пов'язані з кількісними якіс- ними змінами самих фагоцитів Вони можуть бути як набутими, так і спалко во зумовленими, виявляти себе порушеннями як усіх (при лейкопенії), так і окремих функ цій фагоцитів.
Зменшення кількості фагоцитів, а отже і роз лади фагоцитозу, с прямим наслідком лейкопенії (нейтропенія), причини і механізми розвитку якої аналізуються в главі 30. Слід зазначити, що клінічні прояви розладів фагоцитозу (зменшення ня резистентності до інфекцій) виникають лише за умови, що кількість нейтрофілів крові змен шується більше ніж на 50 % відносно норми.
Численні якісні зміни фагоцитів, залежно від того, яка з функція переважно страж дас, можна розділити на такі групи.
1. Зміни рецепторів до хемоатрактантів опсоніни (спадково зумовлені дефекти, блокада рецепторів імунними комплекса ми, як, наприклад, при ревматоїдному ар триті).
2. Дефекти лейкоцитарних молекул клітин ноf адгезії (див, главу 17) та специфічного мембранного глікопротеїну gpl0, унаслі док чого втрачається здатність лейкоцитів взасмодіяти з ендотелієм кровоносних су дин (а отже, і виходити з крові в тканини), а також адгезивність мембрани фагоцитів — неодмінна умова для подальшого перемі щення клітин у тканині (хемотаксису).
3. Порушення мікрофіламентів синдром
"ливих лейкоцитів". Характеризується спадково зумовленою нездатністю актину до полімеризації, унаслідок чого порушу ются міграція процес поглинання лейко- цитами об'єкта фагоцитозу. В експеримент ті моделюється за допомогою антибіотика штохалазину В, які руйнує мікроріла MCHTH.
4. Порушення мікротрубочок — синдром Че дівка – Хігасі. Для нього характерною поява гігантських лізосом, що втрачають здатність зливатися з фагосомами і форму вати фаголізосоми. Крім цього, він виявляє себе порушеннями хемотаксису і секретар ної дегрануляції лейкоцитів. Вважають, що причиною синдрому с спадково зумовлений дефект так званого "великого цитозольного білка", що має стосунок до функцій мікро трубочок і переміщення везикул всередині клітин. В експерименті порушення цього типу відтворюють за допомогою колхицин ну та вінбластину-речовин, що блокують агрегацію тубуліну. У результаті втрача сться здатність лейкоцитів до хемотаксису, але зберігається загальна рухова активність (безладна міграція).
5. Зменшення бактерицидность лейкоцитів.
Описано такі спадково зумовлені розлади:
а) дефіцит НАДФН-оксидази - хронічний гранулематоз. Розвивається у дітей і ха характеризується множинними мікроаб сцесами під шкірою з наступним рубцю ванням і утворенням щільних гранульом.
в основі процесу лежить порушення генерування супероксидного радикалу в НАДФН-оксидазній реакції (див. вище);
б) дефіцит глюкозо-6-фосфатдегідрогенази.
Порушення пентозного циклу, що настає ють при цьому, ведуть до зменшення утво рення НАДФ і до вторинного зниження активності НАДФН-оксидазної системи, в) дефіцит мієлопероксидази. Виявляє себе порушеннями галогенування мембран мікробних клітин.
6. Дефекти лізосомних ферментів. Вони ве дуть до порушень перетравлення різних компонентів убитих бактерій. Результатом цього е незавершени
7. Розлади енергопостачання фагоцитів. За цих умов порушуються процеси, що по требують витрат енергії, а саме: безладна міграція, хемотаксис, поглинання об'єктів фагоцитозу, секреторна дегрануляція.
Оскільки основним джерелом енергозабез печення лейкоцитів с гліколіз, то порушень ня цього типу можна моделювати, викорис товуючи інгібітори гліколітичних фермен тів, зокрема монойодоцтову кислоту.
35. Характеристика первинних імунодефіцитів, які пов’язані з дефектами фагоцитозу
Причини
1 Ренні мутації)Дефектні гени, що утвори лися внаслідок мутацій, передаються або зчеплено зі статтю (1/3 відомих на сьогодні первинних імунодефіцитів), або аутосом но-рецесивно.---- Хромосомні мутації. Найчастіше до розвитку імунодефіцитів ведуть аномалії 1418209 пар хромосом синдром Дауна.
2. Імунологічна недостатність при цьому по єднується з іншими складними синдрома ми, що виникають внаслідок хромосомних аберацій.
3 Кутрішньо Утробні інфекції Часто до виникнення імунологічної недостатності спричиняються вірус червоної висипки Краснуха цитомегаловірус — віруси, що викликають складні вади розвитку плода.
Класифікація. Залежно від рівня порушень і місця виникнення дефекту в імунокомпетентні них клітинах виділяють такі види первинних імунодефіцитів: 0) В-клітинну або гуморальну ні;B (2 Т-клітини і, ago" клітині; 3) поєднані Т.
d В-клітинні, або комбіновані
В-клтини імунодефіцити Bonn naoT y perynsTuri nopymenn mpo- цесі уа орення диференціації В-лімфоцитів, a TAKOK renerHHO yMORnemx posin CHrery Імуноглобуліни,
Серед великої кількості pіних варіантів В-клітинних імунодефіцитів найпоширенішим шими найкраще вивченими стак.
1. AraManobyninemin Epymona. Cnaaomet дефект передасться сплено з Х-хромосо мою, а тому ви для себе тільки у хло кін. Сутність порушень полягає в нездат ності пре-В-лімфоцитів диференціюватися в В-лімфоцити. Унаслідок цього в організмі нема В-лімфоцитів, плазматичних клітин , звісна річ. усіх класів імуноглобулінів (IgM, IgG, IgA, IgD, IgE). Водночас система Т-лімфоцитів не страждає.
Причиною таких порушень с мутація гена Bik (міститься в Х-хромосомі), що кодує фер мент протеїнкіназу, відому як тирозиова кіна- за В-лімфоцитів (В-cell tyrosine kinase Bik).
Цей фермент зв'язаний з рецепторним комплекс сом пре-В- та В-лімфоцитів і відіграє важливу pU у передаванні інформації від рецепторів no renernoro anapary KITHHH. BRaaIOTs, що дефіцит вік неможливо вмикання пев них генів, з якими пов'язана диференціація В-клітин. Зокрема показано, що за відсутності Bik усередині пре-В-лімфоцитів накопичуються ажкі ланцюги імуноглобулінів і не утворюють ся легкі. Це робити неможливим формування рецепторів (IgM) і, як наслідок, перетворення пре-В-лімфоцита і В-лімфоцит (саме наявніс то на поверхні антиген специфічних рецепторів В-лімфоцит і відрізняється від своїх попередни ків).
Хвороба, як правило, розвивається у дітей, починаючи з 6-місячного віку, коли майже пов ністю зникають антитіла, отримані від мле pi. Ocnomni T opoann yMOaneni NOcmsipuuu бактеріальними інфекціями (1) Streptococcus pneumoniae (2) Haemophilus influenzae.
(3) Staphylococcus aureus. Lli 3öyumn C upa- ниною фарингітів, синуситів, середнього отиту.
Oponxiry, mieaon - nasx npouecia a op- raaN AOIro pastry, Корекція даного імунодефіциту здійснюється шелениям в організм імуноглобуліни
Загальний варіабельний імунодефіцит (common variable immunodeficiency CVID). Цей вид первинних імунодефіцитів являє собою збірну групу, до якої входять різні порушення В-лінії лімфоцитів в не з'ясованими до кінця механізмами розви CVID By Являє себе гіпогаммаглобулінеміею, що охоплюс всі класи антитіл, і розвивати- ся, на відміну від агаммаглобулінемії Брутона, в осіб обох статей і досить пізно - у дітей, під літків і навіть дорослих у віці 25-30 років.
Ще одна відмінність від гамма глобулін немії Брутона полягає в тому, що кількість В-лімфоцитів у крові і лімфоїдній тканині хворих на CVID майже не відрізняється від норми. Це свідчить про те, що диференціація пре-В-лімфоцитів у В-лімфоцити відбуваєть ся без перешкод, а страждає перетворення В-лімфоцитів у плазматичні клітини, що про дукують антитіла. Майже повна відсутність плазматичних клітин і спричиняється до того, що не синтезуються всі класи імуногло булінів.
Оскільки для диференціація В-лімфоцитів плазматичні клітини потрібна участь Т-хелперів, то можливими с принаймні два ме механізми порушень цього процесу. Перший з них полягає в тому, що В-лімфоцити, через генетич ні дефекти, не здатні сприймати сигнали, що йдуть від Т-хелперів (напр., брак чи якісні змі ни рецепторів до IL-4, IL-5). Другий механізм припускає спадково зумовлені зміни не в самих В-лімфоцитах, а в Т-хелперам, які втрачають здатність здійснювати активацію нормальних В-клітин з подальшим їх перетворенням у плаз матичні клітини.
Оскільки для CVID характерним є зменшен ня в крові всіх класів імуноглобулінів, то клі нічні прояви і недуги схожі на агаммаглобу лінемію Брутона, тобто у хворих розвиваються запальні процеси, зумовлені гноствірними бак- теріями (в основному у повітроносних шляхах і легенях). Відповідно і корекція імунодефіциту здійснюється введенням в організм готових ан THrin.
3. Cesexmuanui depiyum iaynozaobyainia. Bin B ae a pesyrari nopyweHN CHIresy euro 3 nam Kaacio imynoraobyninin. Hai- частіше буває дефіцит IgA, значно рідше - IgM ra IgG.
Вважають, що в основі порушень утворено ня IgA лежать мутації гена, відповідального за синтез важких ланцюгів а, що входять до скла ду молекул IgA.
Оскільки IgA є секреторним типом імуно Глобулінів, то в умовах дефіциту цих антитіл значно зменшуються захисні властивості слизо вих оболонок щодо багатьох мікробів, зокрема гноствірних бактерій. Це зумовлює частий роз виток запалення в органах дихального, шлунко во-кишкового та урогенітального трактів.
Важливо зазначити, що введення ззовні IgA з метою корекції цього імунодефіциту не тільки не допомагає, ай має тяжкі негативні наслідки, бо спричиняється до алергічних реакцій (див.главу 15). Річ у тім, що введені IgA сприйма ються імунною системою як антигени, адже за повної відсутності IgA в організмі до них не формується імунологічна толерантність. Тому на надходження ззовні IgA імунна система від повідає утворенням анти-IgA антитіл з наступ ною реакція "антиген - антитіло”,
4. Гіпер-IgM синдром. Виявляє себе великим вмістом IgM у крові і практично відсутністю інших класів імуноглобулінів - IgG, IgM,
IgE, наслідком чого висока чутливість хворих до повторних гноєтворних інфекцій.
Кількість т. і В-лімфоцитів при цьому залишається в нормі.
Т-клітинні імунодефіцити
Добавление примечан
1. Синдром Ді Джорджі, або аплазія тимуса.
У його основі лежать вади ембріонально го розвитку, пов'язані з порушенням фор мування органів з 3 і 4 глоткових закутків,
Оскільки тканини цих утворень дають по чаток вилочковій і прищитоподібним зало зам, а також частково тканинам серця і ве ликих кровоносних судин, то синдром ха рактеризується трьома основними групами ознак:
а) імунологічною недостатністю виненість або повна відсутність тимуса);
6) тетанією, що виникає як прояв гіпокальці ємії (недорозвиненість або відсутність при щитоподібних залоз); | в) уродженими вадами серця і аорти.
Відсутність тимуса спричиняється до того, що зовсім не утворюються Т-лімфоцити зі своїх попередників (про-Т-лімфоцитів). Отже, імун на відповідь клітинного типу стає неможливою i організм не може захищатися від вірусних, протозойних та грибкових інфекцій. Крім того, неповноцінними стають і механізми гумораль ної імунної відповіді на тимусзалежні антигени ни, оскільки вони потребують участі Т-хелперів (див. вище). Цим, власне, і пояснюється висока вразливість хворих до інфекцій, що їх зумов люють гноєтворні бактерії. Слід зазначити, що повноцінна гуморальна відповідь на тимуснеза лежні антигени при цьому зберігається.
Аплазія тимуса, будучи вадою ембріонально го розвитку, спадково не передаеться.
Відновлення клітинного імунітету досягають трансплантацією вилочкової залози плода. Пе ресадження тимуса, взятого від дітей старше 14 тижнів, не використовують, оскільки розвива ється реакція трансплантат проти хазяїна”.
2. Синдром Незелофа, або алімфоцитоз. Ця спадкова недуга, що передається за авто типом, характери-сумно-рецесивним зується нездатністю незрілих Т-лімфоцитів диференціюватися в CD4(+)- і CD8(+) лімфоцити — клітини, що здійснюють влас не імунні функції.
Мала кількість Т-лімфоцитів у периферич ній крові та їх функціональна неповноцінність спричиняються до порушень імунологічної ре активності клітинного типу і гуморальних імун них реакцій на тимусзалежні антигени
Поєднані Т-і В-клітинні (комбіновані) іму нодефіцити
1. Тяжкий комбінований імунодефіцит (severe combined immunodeficiency disease- SCID).
Цією назвою позначають велику кількість синдромів з різними генетичними дефекта ми, спільною рисою яких є поєднані пору шення гуморальних і клітинних механізмів імунітету. Імунодефіцит виявляє себе по чинаючи з 3-місячного віку розвитком бак теріальних, вірусних, протозойних і гриб- кових інфекцій. Діти рідко доживають до 2-річного віку, якщо не здійснювати транс плантацію червоного кісткового мозку.
Ще донедавна вважали, що причиною SCID є порушення процесів диференціації стов бурових клітин — спільних попередників Т i В-лімфоцитів. Проте виявилося, що частка таких порушень (т. зв. “класичний”, або швей царський тип імунодефіциту) дуже мала, якщо порівнювати з іншими різновидами SCID. Нині до SCID відносять і ті синдроми, які винника Кть унаслідок первинних генетичних дефектів Т-лімфоцитів, що зумовлюють — через відсут ність чи неповноцінність Т-хелперів — вторинні порушення В-лімфоцитів і, як наслідок, реакцій гуморального імунітету.
За типом спадкування розрізняють (A) SCID, зчеплений з Х-хромосомою, і (Б) той, що пере дається аутосомно-рецесивно.
А. Зчеплений з Х-хромосомою SCID. На нього припадає близько 70 % від усіх ви падків даного виду імунодефіциту, Суть генетичного дефекту полягає в мутації гена, що кодує структуру поліпептидний го у-ланцюга, який входить до складу ба гатьох цитокінових рецепторів, зокрема рецепторів no IL-2, IL-4, IL-7, IL-9, IL-11 та IL-15. За відсутності ү-ланцюга порушу ється утворення, а отже і функції, цитокіни нових рецепторів, унаслідок чого клітини не сприймають регуляторні впливи інтерпол лейкінів. Серед останніх особливо важлиге ве значення для активації мітотичного по ділу попередників Т-лімфоцитів має IL-7. 4-
Дефекти рецепторів до нього зумовлюють припинення процесів проліферації про-Т лімфоцитів як наслідок, не утворюють ся Т-клітини. За відсутності Т-хелперів імунна система, незважаючи на нормальну кількість В-лімфоцитів, втрачає здатність
36 .Визначення та класифікаціяпервиннихімунодефіцитів.
Первинні імунодефіцити- називають імунологічну недостатність, що виникла внаслідок уроджених дефектів імунної системи.
Класифікація: 1) В-клітинні, або гуморальні;
2)Т-клітинні, або клітинні;
3)поєднані Т і В-клітинні, або комбіновані;
37. Характеристика комбінованих первинних імунодефіцитів
Виникають у результаті порушень перетворення стовбурової клітини в клітину-попередницю лімфоцитопоезу або в результаті поєднання дефектів В- і Т-ліній лімфоцитів.
До цієїгрупивходять: а) важкийкомбінованийімунодефіцит(швейцарський тип). Генетичний дефект передається аутосомно-рецесивноабозчеплено з Х-хромосомою. ПорушуєтьсяутворенняВ- і Т-лімфоцитів, синтез імуноглобулінів. Хворідітирідкодосягають 2-річного віку;
б) синдром Луї-Барр. Характеризуєтьсяпоєднаннямімунологічноїнедостатності з атаксією (порушеннямикоординаціїрухів) і телеангіектазією (ураженнямидрібнихсудин). Тривалістьжиттяхворихрідкодосягає 20-30 років;
в) синдромВіскотта-Олдрича. Імунологічнанедостатністьсупроводжуєтьсярозвиткомекземи (ураженьшкіри) і тромбоцитопенії. Як і в попередніхвипадках, страждають
38. Характеристика гуморальнихпервиннихімунодефіцитів
Вони виникають у результаті порушень процесів утворення і диференціації В-лімфоцитів, а також генетично зумовлених розладів синтезу імуноглобулінів.
Серед великої кількості варіантів В-клітинних імунодефіцитів найпоширенішими є:
1.АгаммаглобулінеміяБрутона. Спадковий дефект передається з Х-хромосомою, а тому виявляє себе тільки у хлопчиків . Сутність порушень полягає в нездатності пре-В-лімфоцитів диференціюватися в В-лімфоцити. Унаслідок цього в організмі нема В-лімфоцитів , плазматичних клітин і всіх класів імуноглобулінів.
2. Селективний дефіцит імуноглобулінів . Виникає в результаті порушення синтезу одного з п’яти класів імуноглобулінів. Найчастіше буває дефіцит IgA, значно рідше-IgMта IgG. У хворих спостерігаються рецидивуючі інфекційні захворювання у системі дихання, вибіркове ураження слизових оболонок порожнини рота, шлунково-кишкового тракту. Окрім інфекційних уражень можуть виникати алергічні реакції.
3. Загальний варіабельний імунодефіцит. Цей вид первинних імунодефіцитів являє собою збірну групу, до якої входять різні порушення В-лінії лімфоцитів із не з’ясованих до кінця механізмами розвитку.
39Характеристика клітинних первинних імунодефіцитів.
Складають 5-10% від загальної кількості імунодефіцитів.
1.Синдром Ді Джорджі. Виникає внаслідок порушення ембріонального розвитку тимусу, щитовидної і при щитоподібних залоз. Відсутність тимуса спричиняється до того, що зовсім не утворюються Т-лімфоцити зі своїх попередників.
2.Синдром Незелофа,або алімфоцитоз . Зустічається дуже рідко.Спадковий дефект передається за аутосомно-рецесивним типом. Характерезуєтьсялімфопенією, значним зменшенням Т-клітин при нормальному співвідношенні Т-клітин і Т-супресорів. Порушуються клітинні механізми імунної відповіді.
40. Загальна характеристика порушень діяльності імунної системи: ненормальна імунна відповідь на екзоантигени і втрата толерантності до аутоантигенів. Механізмитолерантностіімунноїсистеми до аутоантигенів. Причини та наслідкиїїскасування.
Порушення діяльності імунної системи, виявляє себе імунологічною недостатністю , і аутоалергічними реакціями.
Імунна відповідь організму на дію антигена здійснюється лімфоїдноюсистемою організму і характеризується:
А) специфічністю (діє на конкретний антиген)
Б) потенціювання (посилення дії при повторному введенні антигена)
В) імунологічною пам’яттю (розпізнає антиген через певний проміжок часу)
Механізми толерантності функціонують постійно, вони необхідні для запобігання розвитку імунної відповіді на нешкідливі антигени .Толерантність може формуватись у Т і В-лімфоцитів .Т-клітинна толерантність розвивається у Т-хелперів, але регулюють відповідь на білкові антигени.
Причини: злоякісні пухлини, порушення харчування, природні імунодефіцити.
Наслідки скасування:аутоімунні хвороби,і гіперчутливість до екзоантигенів.
41. Види імунної недостатності. Етіологія, патогенез первинних і вторинних імунодефіцитів. Типові прояви імунної недостатності у дитячому віці.
Розлади функцій імунної системи можуть проявлятись гіпер-, дис- та гіпофункцією, зміною толерантності антигенів. Гіперфункція імунної системи розвивається при перенапруженні системи антигеном, спадковими змінами синтезу імуноглобулінів, зменшення регуляторних впливів гальмування всередині імунної системи – зниження функції супресорів, а також іззовні – недостатність функції гіпоталамо-гіпофізарно-наднирникової системи. При розвитку пухлин спостерігається гіперфункція імунної системи. При гіперфункції створюються умови для розвитку алергії. Дисфункція імунної системи розвивається при зниженні функції Т-лімфоцитів, що призводять до зниження стійкості організму проти інфекцій. При дефіциті Т-супрессорів, збільшена реакція В-лімфоцитів і вироблення антитіл (IgE) → алергічні реакції. Гіпофункція поділяється на імунодефіцитні (спадкові) і імунодепресивні (набуті). Види первинних імунодефіцитів: 1) пов’язані зі змінами в системі Т-лімфоцитів; 2) пов’язані зі змінами в системі В-лімфоцитів.
Вториною називають набуту імунологічну недостатність.Причинами її можуть бути екзогенні фактори фізичного(іонізуюче випромінювання),хімічні речовини(речи вони,що мають цитотоксичну дію-імунодепресанти) і біологічного(віруси)походження.Ендогеними факторами,що сприяють розвитку вториних імунодефіцитів є старіння,інтоксикації9при уремії,опікова хвороба,злоякісні пухлини) порушення органогенезу імунної системи (в ембріональному періоді); - розлад формування і підтримання імунологічної толерантності (втрата толерантності до своїх антигенів чи набуття толерантності до чужорідних); - порушення ґенезу і функції імуноцитів, спричинені різними етіологічними факторами (канцерогенні, мутагенні, цитостатичні фактори, дефіцит білків та вітамінів, стрес, стероїдні гормони); - розлад внутрішньосистемної гормональної регуляції імунної системи (призводить до зміни стану та ґенезу імуноцитів і недостатності імунної відповіді); - автоімунні хвороби; - пухлина; - інфекційні хвороби; - СНІД.
Типові прояви імунної недостатності у дитячому віці.
Тяжкий комбінований імунодефіцит (ТКІД) є рідкісним синдромом, що може викликатися різноманітними генетичними порушеннями. Цей синдром обумовлений різноманітними генетичними факторами, та поєднаною відсутністю функцій Т- і В-лімфоцитів (а в багатьох випадках також відсутність функції звичайних кіллерів або NK-лімфоцитів). Ці порушення призводять до надзвичайної чутливості до важких інфекцій. В теперішній час відомо дванадцять генетичних причин ТКІД.
Недостатність загального гамма-ланцюга 6 різних цитокінів - Найчастіша форма ТКІД, на частку яких припадає приблизно 45% усіх випадків, обумовлена мутацією гена в Х-хромосомі, що кодує компонент (або ланцюг), який є в рецепторі фактора росту Т-клітин, а також в рецепторах інших факторів росту.
Недостатність аденозиндезамінази - Інший вид ТКІД викликається мутаціями гена, що кодує фермент аденозиндезаміназу (АДА).
Недостатність альфа-ланцюга рецептора ІЛ-17 - Це форма ТКІД обумовлена мутаціями гена 5-ї хромосоми, яка кодує інший компонент рецептора фактора росту – альфа-ланцюг рецептора інтерлейкіна-7 (ІL-7R ).
Недостатність Янус-кінази 3 - Один з видів ТКІД викликається мутаціями гена 19-ої хромосоми, що кодує фермент лімфоцитів Янус-кіназу 3 (Jak3).
Недостатність ланцюгів CD3 - Три інші форми ТКІД, викликані мутаціями генів, що кодують три окремих білкових ланцюги, складові СD3- інший компонент рецепторного комплекса Т-клітин.
Недостатність СD45 - Інший тип ТКІД, обумовлений мутаціями гена, що кодує CD45- білок.
42. Етіологія, патогенез СНІДу. Патофізіологічна характеристика періодів ВІЛ-інфекції. Типові клінічні прояви. Принципи профілактики і терапії ВІЛ-інфекції.
СНІД-інфекційне захворювання,що виникає в результаті ураження вірусом імунної і інших систем,внаслідок чого організм стає високочутливим до вториних інфекцій і злоякісним пухлинам.Причиною Сніда є лімфотропний ретровірус-вірус імунодефіцита людини(ВІЛ).Патогенез.ВІЛ уражає клітини,які на своїй поверхні плазматичної мембрани мають особливий білок СD4+.Цей білок виконує роль рецептора до білка-глікопротеїна вірусної оболонки gp-120.Найбільш чутливими до ВІлує СD4+ Т-лімфлцити.(Т-хелпери).Імунологічна недостатність в умовах Сніду характеризується перш за все недостатність Т-хелперів.
В основі її лежать наступні механізми:1)Руйнування Т-хелперів вірусними частинками,що залишають клітину після репродукції;2)Утворення багатошарового синситія,в якому містяться ураженні вірусом Т-хелпери;30Знищення заражених Т-хелперів цитотоксичними Т-лімфоцитами(Т-кілерами);4)Знищення Т-кілерами незаражених Т-хелперві,до поверхні яких приєднуються вірусні білки.наслідком Недостатності Т-хелперів є:а)Зменшення утворення В-лімфоцитів у відповідь на дію тимусзалежних антигенів,б)порушення активації Т-кілерів,в)Порушення регуляції імуної відповіді в зв’язку зі зменшенням утворення лімфокінів(інтерлейкін-2)
43. Особливості перебігу ВІЧ-інфекції у дітей.
У частини ВІЛ-інфікованих дітей з перинатальним шляхом зараження клінічні прояви виникають рано, захворювання швидко прогресує на першому році життя. У частини ВІЛ-інфікованих дітей симптоми СНІДу не проявляються до шкільного чи навіть підліткового віку.
Період новонародженості
У ВІЛ-інфікованих жінок діти частіше народжуються недоношеними і (або) із затримкою внутрішньоутробного розвитку, тобто з нижчою масою тіла, ніж у неінфікованих жінок.
У дітей, народжених ВІЛ-інфікованими жінками, частіше можуть спостерігатися інші інфекції, зараження якими відбулося в перинатальний період (сифіліс, гепатит, герпес-інфекція, цитомегаловірусна інфекція та ін).
Збільшення лімфатичних вузлів
Збільшення периферичних лімфатичних вузлів – є одним з ранніх симптомів ВІЛ-інфекції у дітей.
Основні ознаки генералізованої лімфаденопатії при ВІЛ-інфекції:
Збільшення одного чи більше периферичних лімфатичних вузлів розміром приблизно 0,5-1см у двох групах чи білатерально в одній групі;
Лімфатичні вузли безболісні при пальпації; не спаяні з навколишніми тканинами, шкіра над ними звичайного кольору і температури;
Збільшення лімфовузлів носить стійкий характер, триває 3 місяці та більше і не пов’язано з гострими запальними процесами.
Збільшення печінки і селезінки – ранній симптом ВІЛ-інфекції, що найчастіше спостерігається. Це пов’язано з безпосереднім впливом ВІЛ.
Порушення темпів фізичного розвитку . Порушення збільшення маси тіла та зниження темпів росту при ВІЛ-інфекції пов’язано:
1. з частими інфекційними захворюваннями;
2. з підвищеними енергетичними витратами організму;
3. з порушенням всмоктування живильних речовин у кишках;
3. з різними соціальними причинами.
Синдром виснаження при ВІЛ-інфекції належить до діагностичних критеріїв СНІДу і визначається як:
втрата більше 10% маси тіла;
підвищення температури тіла постійно чи інтермітуючого характеру протягом 30 днів і більше;
хронічна діарея (дворазові та більше рідкі випорожнення) протягом 30 днів і більше.
Ураження шкіри. Поряд з частими інфекційними ураженнями шкіри (грибковими, бактеріальними, вірусними) при ВІЛ-інфекції у дітей спостерігаються себорейний чи атопічний дерматит, короста, контагіозний молюск, васкуліт, плямисто-папульозний висип.
Ураження дихальної системи при ВІЛ-інфекції у дітей може бути обумовлено збудниками, а також опортуністичними інфекціями. Опортуністичною інфекцією, що спостерігається найчастіше у дітей, є пневмоцистна пневмонія.
Ураження серцево-судинної системи , зокрема серцева недостатність, найчастіше спостерігається у ВІЛ-інфікованих дітей з клінічними проявами СНІДу і (або) важким ступенем імуносупресії.
Ураження травного тракту . Клінічні прояви:
- зниження апетиту, нудота і блювота;
- хронічна діарея; збільшення живота, обумовлене здуттям кишок і збільшенням розмірів печінки і селезінки; випадання прямої кишки.
Нефропатія. Клінічними ознаками нефропатії є: протеїнурія; нефротичний синдром; ниркова недостатність. Ураження центральної нервової системи
Ураження ЦНС зустрічається більш ніж у половини дітей у стадії СНІДу. Причинами є:
1. безпосередній вплив ВІЛ на клітини нервової системи, що розвивається; 2. опортуністичні інфекції і пухлини;
3.токсичний вплив медикаментів;.
Зміни в аналізі крові. У більшості дітей при ВІЛ-інфекції відзначають зміни в загальному аналізі крові: анемія, лейкопенія, тромбоцитопенія.
Опортуністичні інфекції. Серед опортуністичних інфекцій у дітей найчастіше спостерігаються: пневмоцистна пневмонія, мікози, бактеріальні інфекції, у тому числі туберкульоз і атипові мікобактеріози; захворювання, обумовлені групою герпес-вірусів; паразитози (токсоплазмоз, криптоспоридіоз).
Для ВІЛ-інфікованих дітей характерні часті гострі респіраторні вірусні інфекції, важкі бактеріальні інфекції з тенденцією до затяжного, рецидивуючого перебігу і генералізації.
Бактеріальні інфекції у ВІЛ-інфікованих дітей перебігають важко, зі схильністю до рецидивування. Найчастіше спостерігаються гнійний отит, синусит, менінгіт, пневмоніі.
Пухлини. Пухлинні процеси у ВІЛ-інфікованих дітей виникають рідко. Найбільший ризик виникнення новоутворення у 3 стадії захворювання (стадія СНІДу). Основними причинами розвитку пухлин є:
1.дефіцит імунних факторів, що контролюють розвиток пухлин;
2. канцерогенний вплив вірусу Епштейна-Барр, вірусу герпесу 8-го типу;
3.вплив лікарських препаратів, які використовувалися протягом внутрішньо утробного і неонатального періодів.
44. Класифікація імунних реакцій за механізмами пошкодження клітин або їх дисфункції (за Кумбсом і Джелом).
Комплекс порушень, які виникають в організмі внаслідок гуморальних і клітинних імунних реакцій. Класифікація: - за Кумбсом та Джеллом (анафілактичні, реакцій цитолізу, реакції типу феномена Артюса, реакція гіперчутливості сповільненого типу, стимулювальні та гальмівні алергічні реакції); - за часом розвитку: - негайного типу (імуноглобуліни поширюються в організмі, виходять у тканини і секрети, і у випадку повторного введення антигену негайно вступають у реакцію антиген-антитіло); - сповільненого типу (розвивається після повторного надходження антигена в тканини, коли утворюються переважно Т-лімфоцити.
Алергічні реакції 1 типу за Кумбсом та Джелом (анафілактичні).
Тип I — реагіновий (анафілактичний). Антитіла сорбовані на клітині, а антигени поступають ззовні. Комплекси антиген—антитіло утворюються на клітинах, що несуть антитіла. У патогенезі реакцій істотною є взаємодія антигена з IgE і IgG, (реагинамі), сорбованими на тканинних базофілах, і подальша дегрануляція цих клітин. Система комплементу при цьому не активується.медіатори:а)гістамін,б)гепарин,в)повільно реагуюча субстанція анафілаксії,г)фактор еміграції еозинофілів,д)фактор еміграції нейтрофілів,е)фактор активації тромбоцитів,ж)ферменти(нейтральні і кислі протеази)
До цього типу реакцій відносять анафілаксію загальну і місцеву. Загальна анафілаксія буває при анафілактичному шоці. Місцева анафілаксія підрозділяється на: анафілаксію в шкірі (кропив'янка, феномен Овері) і анафілаксію в інших органах (бронхіальна астма, сінна лихоманка).
Алергічні реакції 2 типу за Кумбсом та Джелом. Характеристика стадій.
Антигени знаходяться на поверхні клітин,антитіла в вільному стані.Реакція антиген+антитіло виникає на поверхні клітини.З метою виконати алергічні реакції 2-го типу використовують цитотоксичні сироватки,яка містить антитіла проти антигенів тої чи іншої тканини.Такі сироватки отримуються шляхом імунізації тварин тканиними антигенами.Цитотоксичні реакції проявляються в наступних клінічних формах:ауто алергічні реакції,гематранфузний шок,що виникає при переливанні крові несумісної по групам АВО чи резус-фактор крові,гемолітична хвороба новонароджених,алергія на ліки.
Антигени,що приймають участь в цитотоксичних реакціях:а)компоненти мембран власних клітин(незміні і змінені під дією різних факторів),б)вторино фіксовані на клітиних мембранах антигени(НП лікарські печовини),в)неклітині компоненти тканин(колаген,мієлін)Антитіла:Ig G.По походженю вони можуть бути природні(ізоантитіла),та імуні(утв.при контакті з антигеном).
Для розвитку цитотоксичних реакцій маюь значення наступні властивості антитіл:1)Властивість зв’язувати комплемент,2)ефект опсонізації,3)здатність проникати в тканини.
Виділяють наступні механізми цитоліза:1)Комплемент залежний цитоліз.Після того як антитіла зв’язуються з антигеном на поверхні клітин,виникає фіксація комплемента до Fc-фрагмента імуноглобулінів.В результаті активації комплемента мембрана антигененесучої клітини перфорується і клітина пошкоджується,а потім гине.2)Антитіло залежний фагоцитоз.звязані з антигеном антитіла викликають ефект опсонізації,тобто полегшують фагоцитоз антигеннесучої клітини макрофагами.3)Антитілозалезна клітина цитотоксичність.Знищення антигеннесучої клітини,зв’язаної з антитілами.виконується Т-кілерами.Для патохімічної стадії характерно:
1)Активація комплемента і утворення проміжних (С3b)і побічних (С3а,С5а) продуктів його активації,2)Генерація вільнихрадикалів і пероксидівактивованими макрофагами і Т-кілерами,3)Звільнення макрофагами лізосомальних ферментів.
Патофізіологічна стадія:активований комплемент,вільні радикали,пероксиди і лізосомальні ферменти пошкоджують клітину і викликають її загибель, це призводить до запалення.Якщо кількість антитіл,що реагує з антигеном невелике і являє недостатнім для пошкодження,то виникає ефект стимуляції функціональної активності клітин.
Алергічніреакції 3 типу за Кумбсом та Джелом.
Антиген і антитіло знаходяться у вільному стані.Їх взаємодія знаходиться в крові і тканиній речовині.Імунокомплексні механізми мають значення в патогенезі наступних груп захворювань: 1)Захворюванні обумовлені екзогенними антигенами(сироваткова хвороба,деякі форми алергії на ліки,алергічний альвеолі),2)аутоалергічні хвороби(системна червона вовчанка,ревматоїдний артрит,вузликовий пери артеріїт,тиреоїдит Хашимото),3)Інфекційні захворювання(гепатит В,стрептококова інфекція).Антигени, що викликають розвиток імунокомплексних реакцій повині бути розчиними. Антитіла-Ig G.M.
Пат.властивості циркулюючих імуних комплексів :а)Структурні і функціональні властивості комплексів «антиген-антитіло»,а сааме розміри комплексів і структура їх сітки.,б)тривалість циркуляції імуних комплексів в організмі, в)місцем утворення комплексів.
Патохімічна стадія:1)активаціябіохімічних систем плазмикрові:а)системикомплемента,б)калікреїн-кініновоїсистеми,в)системизгортаннякрові.активація двух останіхпов'язана з пошкодженнямімуними комплексами судиноїстінки,щопризводить до активації фактора Хагемана.2)вивільненняактивованимимакрофагами:а)лізосомальнихферментів,б)основнихкатіоновихбілків,в)вільнихрадикалів і пероксидів.
Патофізіологічна стадія:1)Місцевізміни.Імунікомплексивідкладуються на поверхніендотелія,набазальних мембранах судин,втканинах.Врезультатіактивації комплемента і діїпродуктів ,якідекретують макрофаги виникаєпошкодженняклітин і розвиваєтьсязапалення(глумеролонефрит,альвеолі,васкуліт)Крім того утвореннямікроциркуляції і розвитокнекротичнихзмін в тканинах.2)Загальніпрояви.Активаціябіохімічних систем кровіможе бути причиною ДВС-синдрома.Крім того прикріпленняімунихкомплексів через Fc-рецептори до поверхніформенихелементівкрові,викликаєпоглинання і руйнуванняїхмакровагами.Як результат цитопенія.
Тип III — реакції типу феномену Артюсаабоімуннихкомплексів. Ні антиген, ніантитіло при цьому не є компонентами клітин, і утворення комплексу антиген—антитіловідбувається в крові і міжклітиннійрідині. В даний час налічуєтьсябільше 30 препаратів, якіможутьвикликатисироваткову хворобу, цей синдром схожий з істинноюсироватковою хворобою, щовикликаєтьсягетеролітичноюабогомологічноюсироваткою, виділяютьлегку, важку і анафілактичнуформисироватковоїхвороби. Розвиткугостроїреакціїпередуєінкубаційнийперіод 7-10 дібвід моменту введеннялікарського препарату. Продромальнийперіодхарактеризуєтьсягіперемією, гиперестезієюшкіри, висипаннями в місцяхвведеннялікарськихпрепаратів.
Гострийперіод.Характеризуєтьсяпідвищеннямтемператури до 39-40, поліартралгією, висипом, що сильно зудить. Висипможеноситиерітематозний, папулезний, папуловезикульозний, геммарогічний характер, з'являєтьсяполіартрит, набряк Квінке, нерідкодіагностуєтьсяміокардит, поліневрит, дифузнийгломерулонефрит, гепатит. Гострийперіодпродовжується 5-7 діб, при важкомуперебігу до 2-3 тижнів.
Алергічніреакції 4 типу за Кумбсом та Джелом.
Тип IV — реакціїсповільненоїгіперчутливості (ГЗТ). Головна особливістьреакційсповільненого типу полягає в тому, що з антигеном взаємодіють Т-лімфоцити. Реакціясповільненоїгіперчутливості не меншспецифічна по відношенню до антигена, чимреакція з імуноглобулінами, завдякинаявності у Т-лімфоцитіврецепторів, здатнихспецифічновзаємодіяти з антигеном. Цими рецепторами є, ймовірно, IgM, укорочені і вбудова
45. Вікові особливості механізмів адаптивного (набутого) імунітету у новонароджених та дітей раннього віку.
Із п’яти критичних періодів становлення імунітету у постнатальному періоді, два припадають на перший рік життя (період новонародженості та 3-6-й місяць), що доводить їх важливість. Імунологічний статус новонародженого включає:
пасивний – материнський, що здійснюється за допомогою антитіл класу IgG, а тому забезпечує
протиінфекційний захист;
власне дитини – висока абсолютна кількість Т-лімфоцитів із низькою функціональною зрілістю хелперної та супресорної субпопуляцій, низьким рівнем комплементу, IgA, бактерицидністю фагоцитів тощо, які зумовлюють підвищену вразливість маляти до інфекційних захворювань.
Отже, новонароджена дитина здатна сформувати специфічну імунну відповідь, однак її реакція на первинне вторгнення інфекційних агентів сповільнена, а функціональна можливість Т-лімфоцитів відповідати на стимуляцію обмежена, тому імунна відповідь Т-хелперів зміщується в бік Th2лімфоцитів, чим пояснюється нижча опірність.
Коли дитина досягає 3-6 місячного віку, її імунна система втрачає материнський патронаж, що забезпечувався антитілами класу IgG,– розпочинається синтез власних антитіл імунологічної пам’яті. Послаблення впливу, а згодом втрата пасивного імунітету визначають закономірності роботи імунної системи дитини:
утримується виражений лімфоцитоз;
імунологічні механізми мають супресорну спрямованість;
у відповідь на вторгнення інфекційних агентів збільшується синтез антитіл класу IgМ (лише первинна реакція, яка не забезпечує формування імунологічної пам’яті);
недостатність факторів місцевого захисту.
Загалом, така незрілість системи імунітету визначає підвищену чутливість малюка до збудників ГРВІ (риносинтиціального, аденовірусу, парагрипу) та формування харчової алергії.
Таким чином, перше півріччя життя немовляти характеризується низькою компетентністю імунної системи за умов масивної антигенної агресії ззовні. Саме грудневигодовування є тим еволюційним пристосуванням, що, з одного боку, забезпечує протидію інфекційним збудникам, а з іншого – сприяє дозріванню імунної системи.
46. Вікові особливості механізмів вродженого імунітету у новонароджених та дітей раннього віку
У дітей новонароджених і дітей раннього віку в активному стані знаходяться тільки клітини одного типу, решта знаходиться у фазі сну, тобто, більшість збудників які будуть проникати в організм не будуть виявлені, що призведе до розвитку інфекції в організмі дитини.
47. Визначення поняття “алергія”, принципи класифікації алергічних реакцій. Мультифакторіальна природа алергічних захворювань. Класифікація і характеристика алергенів.
Алергія — це імунна реакція організму, що супроводжується ушкодженням власних тканин. Це якісно змінена реакція організму на дію речовин антигенної природи, яка викликає різні структурні і функціональні порушення.
Основу як алергії, так і імунітету становлять імунні реакції гуморального й клітинного типу, що забезпечують захист проти антигенів. Однак в умовах імунітету знищення антигену відбувається без ушкодження власних тканин, у той час як при алергії таке ушкодження завжди має місце.
Алергени - це речовини антигенної природи, що викликають алергію. Залежно від будови алергени можуть бути повними й неповними (гаптени). Залежно від походження розрізняють екзогенні й ендогенні алергени.
I. За способом проникнення в організм розрізняють:
а) інгаляційні;
б) харчові;
в) контактні;
г) ін'єкційні алергени.
II. За походженням екзогенні алергени бувають:
а) рослинні;
б) тваринні;
в) інфекційні;
г) синтетичні.
Ш. Залежно від джерела надходження в організм виділяють алергени:
а) побутові (домашній пил);
б) промислові (бензол, формалін та ін.);
в) харчові;
г) лікарські;
ґ) пилкові (пилок рослин);
д) епідермальні (шерсть тварин).
Ендогенні алергени (аутоалергени) поділяють на дві групи. І. Природні. Такими є нормальні, незмінені білкові компоненти ряду органів і тканин: мозку, очей, статевих залоз, щитоподібної залози, внутрішнього вуха.
II. Набуті. Це власні білки організму, які змінили свою конформацію внаслідок дії на них факторів зовнішнього середовища. Залежно від природи цих факторів набуті ендоалергени можуть бути неінфекційними (опікові, холодові, променеві) та інфекційними.
Інфекційні набуті ендоалергени — це власні білки організму, змінені під дією інфекційних агентів або продуктів їх життєдіяльності. Вони бувають простими і комплексними (тканина-мікроб, тканина-токсин).
Природні ендоалергени— це компоненти так званих забар' єрних органів, що мають особливим образом улаштовані гістогематичні бар'єри, такі як гематоенцефалічний, гематоофтальмічний, гематотестикуяярний, гематотиреоїдний і гематокохлеарний.
Існування цих бар'єрів унеможливлює контакт імунокомпетентних клітин з антигенами тканин головного мозку, очей, сім'яників, щитоподібної залози, внутрішнього вуха в період ембріогенезу, коли відбувається становлення імунологічної толерантності до власних білків.
Неможливість такого контакту є причиною того,, що до антигенів зазначених вище забар'єрних органів не сформувалася імунологічна толерантність, тобто в організмі існують клони лімфоцитів, здатні давати імунну відповідь на зазначені антигени.
В умовах патології, коли порушується цілісність спеціалізованих гістогематич-них бар'єрів, лімфоцити можуть проникати в забар'єрні тканини і взаємодіяти з нормальними їх компонентами, ініціюючи комплекс клітинних і гуморальних імунних реакцій, - розвивається аутоалергія.
I. За походженням алергенів:
1) алергічні реакції, що спричиняються екзогенними алергенами;
2) аутоалергічні реакції.
II. За клінічними ознаками (класифікація Кука):
1) алергічні реакції негайного типу;
2) алергічні реакції уповільненого типу.
III. За характером і місцем взаємодії алергену з ефекторами імунної системи (класифікація Кумбса і Джслла):
I тип - анафілактичні реакції;
II тип - цитотоксичні реакції;
III тип - імунокомплексні реакції;
IVтип - гіперчутливість уповільненого типу.
IV. За патогенезом:
1) алергічні реакції гуморального типу (І, II і III типи реакцій за Кумбсом і Джеллом);
2) алергічні реакції клітинного типу (IV тип реакцій за Кумбсом і Джеллом).
48. Алергія у дітей. Визначення атопії. Види алергічних реакцій у дитячому віці.
Якщо у обох батьків є алергія, то у дитини ризик захворіти алергією складає 70%. Якщо лише один із батьків хворіє на цю недугу, то до 40%. Якщо ніхто в сім'ї не мав подібної проблеми, то ризик все ж залишається до 20%.
У своїх наукових дослідженнях я займався виявленням генів, які провокують розвиток алергічних хвороб.
Серед видів алергічних реакцій є харчова і нехарчова.
Харчову реакцію може призвести грудне молоко абр прикорм.
Нехарчова алергія
Серед алергенів, не пов'язаних з продуктами харчування, реакцію здатні викликати:
пил і пилові кліщі;
шерсть домашніх тварин;
пилок;
косметичні засоби;
медикаментозні препарати;
в рідкісних випадках холод, укуси комах.
49. Алергічні реакції 1 типу (анафілактичні), за Кумбсом і Джелом. Етіологія, патогенез, клінічні прояви місцевих та системної анафілактичних реакцій. Медіатори анафілаксії. “Псевдо анафілактичні” реакції.
Розрізняють класичний і додатковий механізми патохімічної стадії анафілактичних реакцій.
Сутність класичного механізму полягає в дегрануляції тканинних базофілів, на поверхні яких відбувається реакція антиген+антитіло. У результаті вивільнюються так звані первинні медіатори анафілактичних реакцій. Вони визначають хід подій у перші півгодини після повторного надходження антигену, тобто ранню фазу анафілактичної реакції.
У результаті дегрануляції тканинних базофілів у навкружну тканину виділяються:
а) гістамін (у деяких видів тварин, але не у людини, ще й серотонін);
б) гепарин;
в) повільно реагуюча субстанція анафілаксії (належить до лейкотрієнів);
г) фактор еміграції еозинофілів; ґ) фактор еміграції нейтрофілів;
д) фактор агрегації тромбоцитів;
є) ферменти (нейтральні й кислі протеази).
Значення продуктів дегрануляції тканинних базофілів полягає в тому, що вони безпосередньо діють на клітини-мішені (гладкі м'язи судин, бронхів, матки, кишок,
ендотеліоцити) і втягують в алергічну реакцію інші популяції клітин (еозинофіли, нейтрофіли, тромбоцити).
Ефекти гістаміну в тканинах пов'язані з Н,- і Н2-рецепторами. У низьких концентраціях гістамін стимулює в основному Ht-, у високих — Н2-рецептори.
Основні прояви анафілактичних реакцій зумовлені дією гістаміну на Н,-рецеп-тори. Унаслідок цього виникають такі зміни:
1) скорочення гладких м'язів бронхів, матки, кишок;
2) розширення артеріол;
3) підвищення проникності судинної стінки, в основному на рівні венул;
4) подразнення нервових закінчень (свербіж, біль);
5) збільшення утворення й виділення слизу у верхніх дихальних шляхах.
Дія гістаміну на Н2-рецептори, навпаки, викликає згасання анафілактичних реакцій. Цьому сприяє гальмування дальшої дегрануляції тканинних базофілів, пригнічення активності лімфоцитів, активація Т-супресорів.
1. Активація гістаміном Н,-рецепторів (див. запит. 10.22).
2. Надходження в тканину еозинофілів, які вивільнюють інгібітор дегрануляції тканинних базофілів і ферменти (гістаміназу, арилсульфатазу, фосфоліпазу D), що руйнують первинні медіатори анафілактичних реакцій.
3. Гістамінопексія - зв'язування гістаміну білками сироватки крові.
Цей механізм пов'язаний з активацією не тканинних базофілів, а інших клітин, що мають на своїй поверхні низькоафінні (з низькою спорідненістю) рецептори для фіксації реагінів. Це макрофаги, нейтрофіли, еозинофіли, тромбоцити. їхню активацію викликають великі завершальні дози антигену й продукти дегрануляції тканинних базофілів. Активовані клітини крові вивільнюють речовини, що отримали назву вторинних медіаторів анафілактичних реакцій. Вони обумовлюють розвиток пізньої фази реакцій І типу, ознаки якої виявляються через 6-12 год. і пов'язані з інфільтрацією тканин макрофагами, нейтрофілами й еозинофілами.
Місцеві прояви анафілактичних реакцій пов'язані з дією біологічно активних речовин - продуктів дегрануляції тканинних базофілів. Ці речовини викликають: а) спазм гладких м'язів бронхів - напади ядухи (бронхіальна астма). Розвиваються в результаті дії повільно реагуючої субстанції анафілаксії й гістаміну;
б) алергічний нежить, фарингіт, ларингіт, трахеїт. Ці прояви виникають як наслідок підвищеного утворення й виділення слизу у верхніх дихальних шляхах;
в) спазм гладкої мускулатури кишок - проноси (діарея); w..
г) розширення артеріол - почервоніння, алергічний висип на шкірі, кон 'юнктивіт; ґ) підвищення проникності стінок судин - розвиток місцевих набряків;
д) подразнення нервових закінчень - свербіж, біль.
анафілактичного шоку.
Імунні й патохімічні механізми анафілактичних реакцій в кінцевому підсумку призводять до розвитку функціональних змін, які становлять сутність патофізіологічної стадії алергічної реакції, яка виявляє себе розвитком анафілактичного шоку (рис. 27).
Головними в цей період є такі процеси. І. Порушення загальної гемодинаміки, що виявляють себе насамперед падінням артеріального тиску. В основі цих порушень лежать:
а) генералізоване розширення артеріол і пов'язане з цим падіння загального периферичного опору;
б) генералізоване підвищення проникності судин, що призводить до виходу рідини з судин у тканини і зменшення об'єму циркулюючої крові.
її. Розлади мікроциркуляції. Виникають у результаті падіння артеріального тиску і згущення крові.
III. Розвиток гострої недостатності зовнішнього дихання внаслідок спазму бронхів і бронхіол та закупорки слизом повітроносних шляхів.
Усі зазначені процеси призводять до розвитку гострої гіпоксії, що викликає порушення функції дихального й серцево-судинного центрів і в кінцевому підсумку — смерть.
50. Алергічні реакції 2 типу (цитотоксичні, опосередковані антитілами), за Кумбсом і Джелом. Етіологія, патогенез, клінічні прояви.
Тип ІІ – реакція цитолізу, або цитотоксичної дії, за якої антиген є компонентом клітинних мембран або сорбований на них, а IgMiIgG1,2,3вільно циркулюють у крові. При взаємодії антитіл з антигеном активується комплемент, що призводить до ушкодження клітин. За таким типом відбувається гемоліз еритроцитів при переливанні несумісної крові та при аутоалергічних анеміях, хвороба Верльгофа, реакція відторгнення трансплантату, цитолітична форма гломерулонефриту, дія великих доз антиретикулярної сироватки Богомольця.
51. Алергічніреакції 3 типу (опосередкованіімунними комплексами), за Кумбсом і Джелом. Етіологія, патогенез, клінічні прояви місцевих та системнихреакцій. Сироваткова хвороба.
Третій тип – імунокомплексний (реакції типу феномена Артюса) –.Антиген і антитіло знаходяться у вільному стані.Їх взаємодія знаходиться в крові і тканиній речовині
Імунокомплексні механізми мають значення в патогенезі наступних груп захворювань:1)Захворюванні обумовлені екзогенними антигенами (сироваткова хвороба, деякі форми алергії на ліки, алергічний альвеолі),2)ауто алергічні хвороби (системна червона вовчанка, ревматоїдний артрит, вузликовий пери артеріїт, тиреоїдит Хашимото), 3)Інфекційні захворювання (гепатит В, стрептококова інфекція).Антигени, щовикликають розвиток імунокомплексних реакцій повині бути розчиними.Антитіла-Ig G.M.
Пат.властивостіциркулюючихімунихкомплексів:а)Структурні і функціональнівластивостікомплексів «антиген-антитіло»,а самерозмірикомплексів і структура їхсітки.,б)тривалістьциркуляціїімунихкомплексів в організмі,в)місцемутвореннякомплексів.
Патохімічна стадія:1)активаціябіохімічних систем плазмикрові:а)системикомплемента,б)калікреїн-кініновоїсистеми,в)системизгортаннякрові.активація двух останіхпов'язана з пошкодженнямімуними комплексами судиноїстінки,щопризводить до активації фактора Хагемана.2)вивільненняактивованимимакрофагами:а)лізосомальнихферментів,б)основнихкатіоновихбілків,в)вільнихрадикалів і пероксидів.
Патофізіологічна стадія:1)Місцевізміни.Імунікомплексивідкладуються на поверхніендотелія,набазальних мембранах судин,втканинах.Врезультатіактивації комплемента і діїпродуктів ,якідекретують макрофаги виникаєпошкодженняклітин і розвиваєтьсязапалення(глумеролонефрит,альвеолі,васкуліт)Крім того утвореннямікроциркуляції і розвитокнекротичнихзмін в тканинах.2)Загальніпрояви.Активаціябіохімічних систем кровіможе бути причиною ДВС-синдрома.Крім того прикріпленняімунихкомплексів через Fc-рецептори до поверхніформенихелементівкрові,викликаєпоглинання і руйнуванняїхмакровагами.Як результат цитопенія.
Сироваткова хвороба:
В даний час налічуєтьсябільше 30 препаратів, якіможутьвикликатисироваткову хворобу, цей синдром схожий з істинноюсироватковою хворобою, щовикликаєтьсягетеролітичноюабогомологічноюсироваткою, виділяютьлегку, важку і анафілактичнуформисироватковоїхвороби. Розвиткугостроїреакціїпередуєінкубаційнийперіод 7-10 дібвід моменту введеннялікарського препарату. Продромальнийперіодхарактеризуєтьсягіперемією, гиперестезієюшкіри, висипаннями в місцяхвведеннялікарськихпрепаратів. Гострийперіод. Характеризуєтьсяпідвищеннямтемператури до 39-40, поліартралгією, висипом, що сильно зудить. Висипможеноситиерітематозний, папулезний, папуловезикульозний, геммарогічний характер, з'являєтьсяполіартрит, набряк Квінке, нерідкодіагностуєтьсяміокардит, поліневрит, дифузнийгломерулонефрит, гепатит. Гострийперіодпродовжується 5-7 діб, при важкомуперебігу до 2-3 тижнів.
52. Алергічніреакції 4 типу (опосередкованіклітинами), за Кумбсом і Джелом. Етіологія, патогенез, клінічні прояви.
Тип IV—реакціїсповільненоїгіперчутливості (ГЗТ).
З антигеном взаємодіють Т-лімфоцити. Завдякинаявності у Т-лімфоцитіврецепторів, здатнихспецифічновзаємодіяти з антигеном. Цими рецепторами є, IgM, укорочені і вбудовані в мембрану Т-лімфоцита, і антигенигістосумісності. Проте в тканині, де відбуваєтьсяцяреакція, середбезлічіклітин, руйнуючих антиген і тканину, виявляєтьсялишедекількавідсотків Т-лімфоцитів, здатнихспецифічнореагувати з антигеном. Даний факт став зрозумілийпіслявідкриттялімфокінів — особливихречовин, щовиділяються Т-лімфоцитами. Завдяки ним імунні Т-лімфоцитинавіть в невеликійкількостістаютьорганізаторамируйнування антигену іншими лейкоцитами крові.
53. Алергічні реакції 5 типу (клітинні дисфункції, опосередковані антитілами). Етіологія, патогенез, клінічні прояви.
П'ятий тип алергічних реакцій – рецепторно-опосередкована алергічна реакція негайного типу.
У ролі антигенів при зазначених реакціях виступають нейромедіатори, або гормони (ацетилхолін, інсулін, тиреотропний гормон), що індукують синтез антитіл, головним чином класу IgG, які взаємодіють зі структурами, розташованими в рецепторному комплексі, викликаючи стимулюючий або інгібуючий ефект на клітину-мішень.
При реалізації реакцій цього типу пошкодження клітин не настає, а навпаки – відбувається активація або пригнічення функції клітин. Особливістю цих реакцій є те, що в них беруть участь антитіла, що не володіють комплементзв'язуючою активністю. Якщо такі антитіла спрямовані проти компонентів клітинної поверхні, які беруть участь у фізіологічній активації клітини, наприклад, проти рецепторів фізіологічних медіаторів, то вони будуть викликати стимуляцію даного типу клітин.
54. Визначення псевдоалергічних реакцій та їх відмінності від алергічних. Визначення індивідуальної непереносимості харчових продуктів у дітей.
Псевдоалергічні реакції мають зовнішні клінічні ознаки алергічних, але не є такими, оскільки в їхній основі лежать не імунні механізми (відсутня імунологічна стадія).
Подібні до алергії реакції викликаються:
1) Хімічними факторами, так званими, лібераторамигістаміну, які безпосередньо діють на тканинні базофіли і зумовлюють їх дегрануляцію;
б) порушенням системи комплементу, обумовленим дефіцитом інгібіторів його компонентів або неімунною активацією;
в) порушенням метаболізму поліненасичених жирних кислот, зокрема арахідонової кислоти (аспіринова бронхіальна астма).
Основні принципи попередження і лікування алергії:
1) Запобігання контакту організму з алергенами (етіологічний принцип).
2) Попередження сенсибілізації, коли контакт організму з алергеном є неминучим через: а) створення імунологічної толерантності до даного антигену або б) стану імунологічної супресії, якщо перше є неможливим.
3) Десенсибілізація - введення антигену в сенсибілізований організм дробовими дозами з метою поступового зв’язування антитіл (метод Безредки).
4) Вплив на патохімічну стадію алергійних реакцій: а) попередження утворення і звільнення медіаторів алергії, б) їхня інактивація (наприклад, антигістамінними препаратами); в) блокада рецепторів до медіаторів алергійних реакцій на клітинах-мішенях.
5) Вплив на патофізіологічну стадію - спрямований на ліквідацію структурних і функціональних змін, які виникають при алергії. Це досягається застосуванням протизапальних, спазмолітичних, гіпертензивних та інших фармакологічних середників.
Харчова непереносимість – це імунна реакція організму на різні харчові продути, зумовлена різноманітними порушеннями нормальних процесів травлення та засвоєння їжі.
Харчова непереносимість у дітей може проявлятися з раннього віку та являтися як самостійним порушенням, так і супутнім у комплексі з іншими запальними або функціональними порушеннями органів ШКТ, алергіями, целіакією, лактозною непереносимістю тощо. Імунна реакція на харчові продукти виникає тоді, коли неперетравлені рештки їжі проникають через стінку кишечника у зв'язку з підвищеною проникністю останньої та вступають у контакт з клітинами крові. Існує цілий ряд факторів, асоційованих з розвитком харчової непереносимості, серед яких виділяють наступні:
• генетична схильність;
• незбалансоване харчування та нездорова їжа;
• у дітей харчова непереносимість може виникати внаслідок штучного вигодовування або раннього відлучення від грудного вигодовування;
• промислова обробка продуктів харчування з додаванням великої кількості хімічних добавок (барвники, консерванти, ароматизатори тощо), а також застосування хімічних добрив, пестицидів, антибіотиків, гормонів у сільському господарстві та тваринництві;
• перенесені інфекційні та паразитарні захворювання, алергічні реакції;
• несприятливі екологічні умови;
55. Аутоімунні реакції/хвороби: загальна характеристика, принципи класифікації, сучасні уявлення про етіологію і патогенез.
Аутоімунними реакціями (АІР) називають стани організму, при яких з’являються антитіла чи сенсибілізовані лімфоцити проти нормальних антигенів власного тіла. Такі антигени називають аутоантигенами.
Аутоаллергічні (аутоіммунні) захворювання розвиваються в результаті вироблення антитіл, які можуть взаємодіяти з антигенами власного організму. Це може відбуватися при демаскуванні антигенів, при знятті толерантності і при соматичних мутаціях.
(Демаскування антигенів спостерігається у високодифференційованних органах при порушенні гістогематичних бар'єрів.
Зняття імунної толерантності до нормальних компонентів тканин можливе, наприклад до гаптену свого організму, при заміні носія гаптену.
Соматичні мутації в різних органах можуть призвести до появи клітин, що володіють антигенними властивостями по відношенню до свого організму. )
Мутація імуноцитів може стати причиною аутоіммунних захворювань у зв'язку з тим, що призводить до появи "заборонених" клонів, що сприймають нормальні компоненти організму як антигени.
Встановлено існування ще двох механізмів аутоіммунних захворювань:
а) недостатність антиідіотипічних антитіл;
б) зрив розпізнавання "свого" за допомогою рецепторів, в якості яких Т-лімфоцити використовують антигени головного комплексу гістосумісності МНС. (majorhistocompatibilitycomplex – главный комплекс гистосовместимости).
Залежно від локалізації патологічного процесу АІЗ поділяють на органоспецифічні і системні (неорганоспецифічні).
При органоспецифічних хворобах патологічний процес має локальний характер і розвивається у певному органі. Органами-мішенями при органоспецифічних АІЗ стають частіше щитовидна залоза, шлунок, підшлункова та надниркові залози. При системних хворобах, в тому числі ревматологічних, звичайно уражуються шкіра, нирки, сустави і м’язи.
При системних (неорганоспецифічних) АІЗаутоантигени реагують з різними тканинами даного чи навіть іншого виду тварин. Аутоантигени в цьому випадку не ізольовані від контакту з лимфоїдними клітинами, і аутоімунізація розвивається внаслідок зриву існуючої раніше толерантності. Експериментально відтворення системної аутоімунізації на тваринах ускладнено, але в деяких випадках захворювання може розвиватися спонтанно. Типовими системними аутоімунними хворобами є: системна червона вовчанка, дискоїднаеритематознавовчанка, дерматоміозит, склеродермія, ревматоїдний артрит. Системні АІЗ частіше виникають у людей похилого віку.
До змішаних АІЗ можна віднести: виразковий коліт, первинний біліарний цироз, міастенію гравіс, аутоімунну гемолітичну анемію, синдром Сьйогрена, ідіопатичнутромбопенічну пурпуру, ідіопатичну лейкопенію тощо.
56.Основи трансплантації органів і тканин. Причини і механізми відторгнення трансплантату, засоби попередження. Реакції “трансплантат проти хазяїна” (на прикладі пересадки кісткового мозку при лейкозах у дітей).
Трансплантація (від лат. transplantatio — пересаджування) — метод, що полягає в пересадці реципієнту органу або тканини (трансплантата), взятих у донора, а також клонованих тканин, штучних імплантатів (електронних, металічних та інших), найчастіше методом хірургічного втручання.
Основні принципи застосування трансплантації
Трансплантація здійснюється на таких принципах:
надання донорських органів потенційним реципієнтам за медичними показаннями;
безоплатності трансплантації для донора та реципієнта;
Основний спосіб зберігання органів для трансплантації – консервація.
Реакція відторгнення трансплантата - імунна відповідь реципієнта на пересадку
чужорідного органу або тканини (Алотрансплантація). Відноситься до
реакцій трансплантаційного імунітету.
Розрізняють блискавичний (розвивається через хвилини після підключення органу до кровотоку), гостре (0-3 тижні після операції) і хронічне відторгнення (через кілька місяців або пізніше).
Причини відторгнення:
Відторгнення трансплантата пов'язане з дією Т-лімфоцитів, направленою безпосередньо і специфічно проти антигенів донора. Це можуть бути цитотоксичні клітини (Т- хелпери CD4 або Т-супрессори CD8) або клітини, що опосередковують дію ДТН (відносяться до класу Т-лімфоцитів CD4)
Механізми відторгнення алотрансплантатів.
Імунопатологічна реакція відторгнення ґрунтується на відповідних механізмах антитілзалежного чи клітинного ушкодження тканин, чи їхньої комбінації.
У залежності від механізму імунної реакції:
- гуморальне відторгнення – реалізується за допомогою гуморальних антитіл;
- клітинне відторгнення – реалізується за допомогою лімфоцитів.
У залежності від швидкості клінічних проявів:
- зверхгостре відторгнення, при якому трансплантат може бути ушкоджений і цілком зруйнований протягом декількох годин чи навіть хвилин після реваскулярізації;
- гостре відторгнення – ушкодження трансплантата протягом перших трьох тижнів після трансплантації;
- хронічне відторгнення – розвивається протягом декількох місяців чи років. У залежності від важкості клінічних і патоморфологічних змін:
- легке відторгнення; - помірне відторгнення; - важке відторгнення.
Для
зниження ризику подібних реакцій застосовується підбір донора за
антигенами головного комплексу гістосумісності (MHC), також часто по системі
ABO і різні імунодепресанти для пригнічення імунної системи, деякі з яких
можуть призначатися довічно.
Вважається, що для успішної
алотрансплантації органу необхідно збіг реципієнта і донора хоча б по п'яти
з шести основних антигенів MHC. Розбіжність за двома антигенів MHC не виключає
можливість трансплантації в принципі, але сильно підвищує ймовірність реакції
відторгнення трансплантата. Розбіжність за трьома і більше антигенів MHC
виключає саму можливість трансплантації від даного донора даному реципієнту
Для алотрансплантації, навіть при наявності
ідеально сумісного (збіг шість з шести антигенів MHC) і близькоспорідненого
донора, також потрібна висока ступінь імуносупресії (пригнічення імунної
системи) організму реципієнта, з тим, щоб придушити можливе відторгнення трансплантата
(наприклад, через розбіжність мінорних антигенів гістосумісності) і забезпечити
його приживлюваність. В разі неповного збігу по МНС або при неспорідненій
трансплантації вимоги до рівня забезпечується імуносупресії ще вище.
Реакція "трансплантат проти хазяїна" (гомологічна хвороба) — це імунна агресія пересаджених клітин донора, спрямована проти антигенних структур реципієнта.
Якщо
донорський кістковий мозок недостатньо генетично відповідає тканинам
реципієнта, він може сприйняти тканини його організму, як чужорідний матеріал,
атакує і почне руйнувати його. Цей стан відомо як реакція - трансплантат проти
хазяїна
і може бути небезпечною для життя хворого.
57.Характеристика поняття „пошкодження”. Принципи класифікації пошкодження клітин. Структурні, функціональні, фізики-хімічні, біохімічні та термодинамічні ознаки пошкодження клітини. Екзо- і ендогенні причини пошкодження клітин.
Пошкодження клітини (альтерація) - це типовий патологічний процес, основу якого складають порушення внутрішньоклітинного гомеостазу, що приводять до порушення структурної цілісності клітини і її функціональних здатностей.
Ушкодження клітин класифікують:
1) У залежності від швидкості розвитку ушкодження, розрізняють: а) гостре - розвивається швидко, як правило внаслідок одноразового, але інтенсивного впливу ушкоджуючого агента,
б) хронічне - перебігає повільно і є наслідком тривалого, проте менш інтенсивного патогенного впливу.
2) У залежності від ступеня порушень внутрішньоклітинного гомеостазу, розрізняють: а) зворотні - зникають після припинення дії ушкоджуючого фактора, б) незворотні - ведуть до загибелі клітини.
3) В залежності від періоду життєвого циклу клітини: а) мітотичне і б) інтерфазне.
4) В залежності від етіологічного фактора: а) безпосереднє (первинне) ушкодження та б) опосередковане ушкодження.
5) У залежності від патогенетичних механізмів ушкодження клітин поділяють на: а) насильницьке і б) цитопатичне
Про ушкодження клітини свідчать наступні ознаки:
1) Структурні - виявляються за допомогою гістологічних і електронномікроскопічних методів.
2) Функціональні - супроводжуються: а) порушенням електрофізіологічних процесів, б) порушенням скоротливості, в) порушенням екзо- і ендоцитозу; порушенням клітинного поділу і т.ін.
3) Фізико-хімічні – супроводжуються: а) порушенням з боку клітинних колоїдів, б) змінами водно-електролітного обміну і т.ін.
4) Біохімічні – супроводжуються порушенням внутрішньо- і позаклітинної концентрації різноманітних речовин.
5) Термодинамічні – супроводжуються конформаціїними змінами макромолекул, які відбуваються в напрямку найбільш вигідного термодинамічного стану (денатурація).
Екзо- і ендогенні причини пошкодження клітин: гіпоксія, дія фізичних, хімічних, інфекційних агентів, імунні реакції, генетичні дефекти.
Первинно-ушкоджуючі фактори:(фізичні, хімічні, біологічні чинники)
Вторинно-ушкоджуючі фактори: (гіпоксія, гіпо- і гіпертермія, гіпо- і гіперосмія, ацидоз і алкалоз тощо)
58.Характеристика універсальних механізмів пошкодження клітин: О2-залежні (дія кисню та його похідних – вільних радикалів; перекісне окислення ліпідів).
Пероксидним окислюванням ліпідів називається вільнорадикальне окислювання ненасичених жирних кислот, які входять до складу фосфоліпідів клітинних мембран.
Ініціаторами ПОЛ є вільні радикали - це молекули або атоми з непарним числом електронів, що надає їм високої реакційної здатності та можливості взаємодіяти з різними речовинами. Серед них найбільше значення мають: а) •O2ֿ (НО2•) - супероксидний радикал; б) ОН• - гідроксильний радикал; в) Н• - водневий радикал; г) •O2 - синглетний (збуджений) кисень.
Один із таких первинних вільних радикалів (А*) взаємодіє з молекулою ненасиченої жирної кислоти (ЖК), в результаті чого утворюється вільний радикал цієї кислоти (Ж*) і молекулярний продукт реакції (КА):
А* + ЖК = Ж* + КА.
Вільний радикал жирної кислоти (Ж*) дальше взаємодіє з молекулярним киснем (О2), який завжди є у клітині, в результаті чого утворюється пероксидний радикал цієї кислоти:
Ж* + О2 = ЖОО*
Пероксидний радикал (ЖОО*), в свою чергу, взаємодіє із поряд розташованою молекулою ненасиченої жирної кислоти (ЖК), в результаті утворюється гідропероксид (ЖООК) і новий вільний радикал (Ж*):
ЖОО* + ЖК = ЖООК + Ж* .
Слід зазначити 2 важливі особливості ПОЛ:
□ По-перше: реакції ПОЛ мають ланцюговий характер. Це означає, що в ході реакцій ПОЛ не відбувається знищення вільних радикалів і в процес залучаються все нові і нові молекули ненасичених жирних кислот.
□ По-друге: реакції ПОЛ мають розгалужений характер. Іншими словами, у реакціях ПОЛ у зростаючій кількості з’являються вільні радикали, джерелом яких є самі проміжні продукти ПОЛ. Прикладом може бути утворення вільних радикалів з гідропероксидів ліпідів при їх взаємодії з наявними в клітині металами перемінної валентності:
ЖOOH + Fe2+ → ЖO• + ОНֿ + Fe3+.
□ За ходом багатьох нормально перебігаючих біохімічних реакцій в організмі утворюється невелика кількість вільних радикалів. Але у клітині постійно існує небезпека активації ПОЛ. Однак у природних умовах цього не відбувається, оскільки клітина має у своєму розпорядженні механізми антиоксидантного захисту, завдяки яким досягається інактивація вільних радикалів, обмеження і гальмування ПОЛ.
59.Характеристика універсальних механізмів пошкодження клітин: кальцій-залежні механізми пошкодження клітин.
Кальцієві механізми
Ушкодження клітинних структур може бути обумовлено стійким підвищенням концентрації іонів Са2+ у цитоплазмі клітини. Така ситуація виникає: 1) в результаті надлишкового надходження іонів Са2+ у цитоплазму (гіперкальціємія, підвищення проникності плазматичної мембрани), або в результаті порушення механізмів, що забезпечують видалення іонів Са2+ з цитоплазми (порушення Са-насосів, Na-Ca-обмінного механізму, Са-акумулючої функції мітохондрій).
Підвищення концентрації іонів Са2+ у цитоплазмі викликає:
а) контрактуру фібрилярних структур клітини (міофібрил, елементів цитоскелету);
б) активацію фосфоліпази А2;
в) порушення зв’язку між процесами окислювання і фосфорювання.
60.Характеристика універсальних механізмів пошкодження клітин: електролітно-осмотичний і ацидотичний механізми пошкодження.
Електролітно-осмотичні механізм ушкодження клітини зумовлені змінами вмісту основних клітинних катіонів Na+ і K+. Вирівнювання концентрацій іонів Na+ і К+ з обох боків плазматичної мембрани (збільшення вмісту Na+ і зменшення вмісту K+ у цитоплазмі) у своїй основі може мати два механізми: 1) посилення дифузії іонів через плазматичну мембрану по існуючому концентраційному й електричному градієнті і 2) порушення механізмів активного транспорту Na+ і K+ (Na-K-насоса).
Перший механізм реалізується в умовах загальних порушень водно-електролітного обміну (гіпернатріемія, гіпокаліемія) і порушення бар’єрної функції плазматичної мембрани (підвищення її іонної проникності).
Розлади функції Na-K-насоса можуть бути обумовлені дефіцитом АТФ у клітині, збільшенням вмісту холестерину в ліпідному бішар мембрани (наприклад, при атеросклерозі), дією цілого ряду специфічних інгібіторів Na-K-АТФ-ази (наприклад, строфантину).
Зрушення у вмісті іонів Na+ і К+ викликають: а) втрату клітиною електричного мембранного потенціалу (потенціалу спокою); б) набряк клітини; в) осмотичного розтягання клітинних мембран, що супроводжується підвищенням їх проникності.
Ацидотичний механізми пошкодження.
До розвитку внутрішньоклітинного ацидозу можуть приводити:
1) надлишкове надходження іонів Н+ у клітину з позаклітинного середовища (декомпенсований газовий чи негазовий ацидоз);
2) надлишкове утворення кислих продуктів у самій клітині при активації гліколізу (молочна кислота), порушеннях циклу Кребса (три- і дикарбонові кислоти), гідролітичному розщепленні фосфоліпідів клітинних мембран (вільні жирні кислоти, фосфорна кислота) і ін.;
3) порушення зв’язування вільних іонів H+ у результаті недостатності буферних систем клітини;
4) порушення виведення іонів H+ із клітини при розладах Na-H-обмінного механізму, а також в умовах порушеного місцевого кровообігу в тканині.
Внутрішньоклітинний ацидоз викликає:
а) зміну конформації білкових молекул з порушенням їх ферментативної, скорочувальної і іншої властивостей;
б) підвищення проникності клітинних мембран;
в) активацію лізосомальних гідролітичних ферментів.
61.Механізми пошкодження
клітин викликані активацією протеолізу, денатурацією білків та генетичного
апарату клітини.
Активаціяпротеолізу –
відбуваєтьсяпіддієюлізосомнихгідролітичнихферментів (катепсинів), наприклад,
при запаленні і Са-активованих протеаз
Денатцраціябілків- порушення нативної будови білкових молекул внаслідок
змін вторинної і третинної структури білка, зумовлених розривом нековалентних
зв’язків (денатурація білків сироватки крові при дії етанолу, сечовини,
меркаптоетанолу, вплив іонізуючого випромінювання, підвищеної температури);
62.Некроз та
апоптоз, їх характерні ознаки. Екзо- та ендогенні індуктори апоптозу. Шляхи та
механізми апоптозу.
Некроз-загибель необоротно ушкоджених клітин ,яка відбувається за
участю лізосомних ферментів,що здійснюють гідролітичне розщеплення усіх
компонентів клітин ,руйнують плазматичну мембрану і зумовлють вихід вмісту
клітини за її межі в навкружну тканину знаступним розвитком запалення .
Апоптоз–запрограмована загибель клітини ,що відбувається за участю спеціально
призначених для цього активованих ферментів ,які руйнують ДНК ,ядерні білки і
білки цитоскелета ,не ушкоджуючи при цьому плазматичну мембрану .
Три фази:
1)ініціювання –суть її полягає в послідовності активації каспаз
є три шляхи ініціювання апоптозу :
а)рецептор-опосередкованний0
б)мітохондріальний-відбувається вихід проапоптичних білків з мітохондрій у
цитоплазму в умовах підвищення проникності мітохондріальних мембран
в)ядерний –відбувається ушкодження генетичного матеріалу
2)вбивання –утв.активні форми кас паз-екзекуторів-- ці протеази :розщеплюють
білки цитоскелета ,гідролізують білки матриксу ядра --унаслідок змін. Форма і
об’єм клітини ,ядро розпадається на окремі фрагменти
3)вилучення загиблих клітин (фагоцитоз)-здійсн.макрофагами шляхом фагоцитозу
мертвих клітин .важливе значення циого етапу є своєчасний фагоцитоз загиблих
клітин запобігає їхньому некрозу ,виходу з клітин лізосомних ферментів і
розвитку запалення
63.Запалення:
визначення поняття, принципи класифікації. Характеристика гострого та
хронічногозапалення. Загальні прояви та місцеві ознаки запалення. Етіологія
запалення.
Запалення-типовийпатологічнийпроцес
,щовиникає в результатіушкодженнятканини й виявляє себе комплексом структурних
,функціональних та метаболічнихпорушень ,а такожметаболічнихпорушень ,та
розладамимікроциркуляції.
Запаленнякласифікується за:
· виразністю основного місцевогопроцесу: альтеративне, ексудативне (за видом ексудату: серозне, гнійне, геморагічне, фібринозне, змішане) і проліферативне;
· реактивністюорганізму: нормергічне, гіпоергічне і гіперергічне;
·
перебігом: гостре,
підгостре, хронічне.
Характеристика гострого та хронічного запалення:
Сутністюгострогозапалення є
повнезнешкодження та елімінаціяпатологічного агента з осередкузапалення і
завершенняпатологічногопроцесуповнимвідновленнямцілісностіпошкоджених тканин.
Хронічнезапаленнярозвивається як результат постійного,
тривалоговпливупатологічного агента на орган, тканину,
тобтоорганізмнеспроможнийзнищити та видалитичинникзапалення.
прояви та ознаки:припухлість ,почервоніння ,жар,біль,порушення функції
Етіологія:
Серед зовнішніх чинників запалення виділяють фізичні (чужорідні тіла, сильний
тиск на тканину, висока чи низька температура, іонізуюче і ультрафіолетове
проміння, високий і низький барометричний тиск, електричний струм), хімічні
(кислоти, луги, солі важких металів), біологічні (бактерії, віруси, грибки,
черви, комахи). До внутрішніх чинників запалення належать ті агенти, що
виникають в організмі внаслідок якогось захворювання, наприклад жовчні кислоти,
комплекс антиген-антитіло та інші.
64.Патогенез
гострого запалення, стадії. Поєднання патологічних та
пристосувально-компенсаторних змін в динаміці гострого запалення. Альтерація,
види, причини, механізми.
1)альтерація
2)порушення мікроциркуляції з явищами ексудації та еміграції
3)проліферація
У основі альтерації лежать дві групи явищ: 1) пошкодження кліток і
позаклітинних структур; 2) утворення медіаторів запалення.Первиннаальтерация—
це пошкодження тканини, що виникає унаслідок безпосередньої дії флогогенних
агентів. Вторинна альтерация— це пошкодження тканини, що виникає в
результаті дії чинників, які утворилися унаслідок первинної альтерації. . чинники
,що викликають розвиток вторинної альтерації у вогнищі запалення: 1.
Медіатори запалення (лізосомальні чинники, активований комплемент, лімфокини-лімфотоксини).
2. Вільні радикали і пероксиди. 3. Гіпоксія, що виникає в результаті місцевих
розладів кровообігу. 4. Місцевий ацидоз. 5. Підвищення осмотичного і
онкотичного тиску у вогнищі запалення.механізми ,що,лежать в основі розвитку
місцевих клінічних ознак запалення: 1. Почервоніння (rubor) пов'язане
з розвитком артеріальною, а потім і венозній гіперемії. 2. Підвищення місцевої
температури (calof) обумовлене підвищенням інтенсивності катаболічних процесів
у вогнищі запалення, а також артеріальною гіперемією, під час якої в тканину
поступає багато теплої артеріальної крові.
65.Зміни фізико-хімічних властивостей у вогнищі гострого запалення. Ацидоз: характеристика та стадії. Тканинна гіперплетія. Зміни колоїдного стану.
Зміни фізико-хімічних властивостей у
вогнищі гострого запалення:
1)ацидоз
2)гіперосмія
3)гіперонкія
Розрізняють первинний і вторинний ацидоз .Первинний ацидоз виникає в перші 30хв
. унаслідок де полімеризації основної інтерстиціальної речовини і вивільнення
карбоксильних і сульфатних груп .
Вторинний ацидоз розвивається пізніше й обумовлений порушенням обміну молочної
кислоти ,вихід з ушкоджених клітин недоокисненних продуктів циклу Кребса
,вивільнення вільних жирних кислот,амінокислот і фосфорнох кислоти в
результаті гідролітичного розщеплення тригліцеридів ,фосфоліпідів ,білків та
АТФ.
Гіперплазія – збільшення об’єму ( і маси) органа\тк.
за рахунок збільш кіл-ті кл.,що входить до складу органа.
66.Порушення обміну речовин в осередку запалення.Порушення обміну білків, жирів , вуглеводів та енергетичного гомеостазу .
Запалення починається з посилення обміну речовин,
надалі послаблюється. В гострий період переважають катаболічні процеси
(деполімеризація білково-глікозоаміногліканових комплексі, розпад білків,
жирів, вуглеводів, знаходять вільні АК, поліпептиди, аміноцукри, уронові
кислоти).
Анаболічні процеси проявляються на ранніх стадіях, але переважають в більш
пізніших стадія, коли виникають репаративні процеси (посил.синтез ДНК і РНК,
зростає активність гістіоцитів і фібробластів, в яких активуються процеси
окиснення і окисного фосфорилювання, посил.утвореннямакроергенів).
Ферменти, які вивільнилися з пошкоджених лізосом, гідролізують вуглеводи, білки, НК, жири у вогнищі запалення. Продукти гліколізу піддаються дії ферментів гліколізу, при цьому споживання кисню посил.на 30% (триває 2—3год). Надалі альтерація клітин супроводжується пошкодженням мітохондрій, в таких умовах окислення ще більше порушується в умовах незміненого гліколізу -> збільшується рівень молочної кислоти і трикарбонових кислот -> окислення в циклі Кребса не заверш. ->пригніч.утворення вуглекислоти -> дихальний коефіцієнт знижується.
67.Медіатори запалення, їх класифікація. Механізми утворення та біологічної дії плазмових медіаторів запалення.
Медіатори запалення– біологічно активні речовини, які синтезуються в клітинах або в рідинах організму і спричинюють безпосередній вплив на запальний процес.
Класифікація:
- клітинного, плазмового походження
- хімічна структура (біогенні аміни, поліпептиди, білки);
- походження (похідні арахідонової кислоти, гуморальні, клітинні);
- механізм (нецитотоксичні, цитотоксичні);
- термін дії (миттєвої, уповільненої);
- характер дії (прямої дії, непрямої дії)
Гуморальні (плазмові) медіатори запалення (кініни та система комплементу) синтезуються в плазмі крові під дією певних ферментів (які в свою чергу активуються лізосомальними ферментами – медіаторами клітинного походження, що з’являються внаслідок альтерації клітин).
Медіатори плазмового походження утворюються поза клітинами - у кровоносних судинах і в інтерстиції - з білків плазми крові.
Біологічний вплив: розширення капілярів, збільшення проникності їх стінки, біль, свербіння, активізація лейкоцитів, хемотаксис, цитоліз).
68.Медіатори запалення клітинного походження; характеристика їх біологічних ефектів. Значення біогенних амінів, простагландинів, лейкотриєнів та відповідні напрямки патогенетичної корекції у дитячому віці.
До медіаторів клітинного походження відносять
лізосомні фактори, продукти вільнорадикального окиснення, продукти тканинних
базофілів, похідні арахідонової кислоти, цитокіни, фактори росту. Медіаторами
Лізосомні факторируйнуючи клітинні та позаклітинні компоненти тканини, спричиняють вторинну альтерацію у вогнищі запалення.
утворення і вивільненняіншихмедіаторів запального процесу, зокрема:
стимулюютьдегрануляціютканиннихбазофілів;
активуютьбіохімічнісистемиплазмикрові (калікреїн-кінінову, системи комплементу, зсіданнякрові і фібринолізу);
гідролізуючифосфоліпідиклітинних мембран, сприяютьвивільненнюполіненасиченихжирових кислот і утворенню з них ліпіднихмедіаторівзапалення
продуктивільнорадикальногоокиснення- здатні ініціювати і посилювати процес вторинної альтерації
продукти тканинних базофілів- мастоцити , лаброцити -Нейропаракринний механізм регуляції діяльності кровоносних судин , Провідна роль в процесі виділення медіаторів запалення
похідні арахідонової кислоти-
простогландини :розширення артеріол і розвиток пов'язаної з цим артеріальної гіперемії;
звуження венул і збільшення гідродинамічного тиску в капілярах, що сприяє розвиткові набряку;
збільшення проникності стінок капілярних судин;
посилення хемотаксису лейкоцитів ;
зменшення порога чутливості больових рецепторів до різних подразників.
лейкотрієни : стимулюють хемотаксис нейтрофілів ; спричиняють звуження артеріол; підвищують проникність стінок кровоносних судин
тромбоксани. Вивільнившись з активованих кров'яних пластинок, тромбоксан A2 спричиняє дві важливі реакції: (1) звуження артеріол і (2) агрегацію тромбоцитів.
Цитокіни-вторинна альтерація , еміграції лейкоцитів. проліферації.загальнихпроявівзапалення, відомих як "реакціягостроїфази".
Біогенні аміни- серотонін і гістамін
1)підвищення проникності мікросуцин (венул) - одна з причин запального набряку;
2)подразнення нервових закінчень, що зумовлює розвиток болю;
3) спазм гладких м'язів бронхів, матки, кишок.
Простагландини утворюються практично у всіх клітинах. Вони мають властивість розширювати артеріоли, звужувати венули, підвищувати проникність судинної стінки, зменшувати поріг больової чутливості нервових закінчень.
Місцем утворення лейкотрієнів є лейкоцити і тканинні базофіли. У вогнищі запалення вони стимулюють хемотаксис лейкоцитів і підвищують проникність кровоносних судин.
В дитячому віці нам треба застосовувати інгібітори і блокатори , тобто глюкокортикоїди бо вони мають портизапальну дію, протиалергічну , зменшують кількість цих клітин які йдуть у вогнище запалення , зменшують розширення судин ,пригнічують фагоцитоз, ( чисто інтуіція і логіка, бо ніде сука немає цього!!)
69. Типові зміни місцевого кровообігу при запаленні (дослід Ю. Конгейма). Патогенез окремих стадій судинної реакції у вогнищі гострого запалення
Ю. Конгейм вперше вивчив в експерименті на жабі кровообіг у брижі під час запалення , встановивши всі його стадії від гіперемії до стазу. Він також описав процес еміграції лейкоцитів крізь судинну стінку. Запропонована ним судинна теорія пояснювала розвиток основних проявів запалення саме порушеннями мікроциркуляції в ушкодженій тканині.
стадії порушень місцевого кровообігу у вогнищі запалення.
I. Короткочасна ішемія (тривалість від 10—20 с до кількох хвилин).
II. Артеріальна гіперемія (триває 20—30 хв, максимум до 1 год).
III. Венозна гіперемія.
IV. Стаз
1. Короткочасна ішемія виникає внаслідок спазму артеріол , триває залежно від сили ушкоджувальногоагента від 3-5 с до декількох хвилин.
Короткочасну ішемію на початку запалення обумовлює рефлекторний спазм артеріол. Збудженням судинозвужувальних адренергічних нервів і виділенням їхніми закінченнями катехоламінів, ВОНИ діючи на альфа- адренорецептори, викликають скорочення гладких м'язів судинної стінки. Ішемія - короткочасна, тому що швидко настає виснаження катехоламінових депо в нервових закінченнях і відбувається руйнування вивільнених медіаторів відповідними ферментами, зокрема, моноаміноксидазою. Крім того, вазоконстрикція в деяких тканинах може перекриватися судинорозширювальним впливом холінергічних нервів, що реалізується за типом аксон-рефлексу.
2. Артеріальна гіперемія
Механізми розвитку артеріальної гіперемії у вогнищі запалення.
1. Нейрогенні механізми (нейротонічний і нейропаралітичний). Відбувається швидке спустошення катехоламіновихпресинаптичних везикул у закінченнях судинозвужувальних нервів з одночасним руйнуванням вивільненого в тканину норадреналіну відповідними ферментами зокрема моноаміноксидазою( нейропаралітичний механізм)
2. Вплив фізично-хімічних факторів: ацидозу, збільшення вмісту іонів калію в тканині, гіпоксії та ін.
3. Вплив продуктів метаболізму: молочної кислоти, АДФ, АМФ, аденозину.
4. Дія медіаторів запалення:
а) продукти дегрануляції тканинних базофілів (гістамін , серотонін)
б) кінінів (брадикініну і калідину);
в) похідні арахідонової кислоти (простагландинів і простациклінів)
3. Венозна гіперемія
І. Внутрішньосудинні фактори:
1) збільшення в'язкості крові -настає внаслідок переходу рідини із судин у запалену тканину в процесі ексудації
2) мікротромбоутворення - зумовлене ушкодженням ендотелію судин та пов'язаними з цим процесами адгезі,ї агрегації тромбоцитів. Агрегацію тромбоцитів за цих умов стимулює чинники тканинні ,плазмові і тромбоцитарні.
3) зсідання крові - започатковує активація фактораХагемана завдяки контакту з ушкодженим ендотелієм
4) крайове стояння лейкоцитів - початковий етап еміграції настає одночасно з розвитком венозної гіперемії. Його суть складають три послідовні події
*Маргінація лейкоцитів перехід білокрівців із циркуляційного в пристінний (маргінальний) пул.. Як тільки уповільнюється течія крові в судинах (венозна гіперемія) лейкоцити починають переходити з осьової зони на периферію.
*Оборотна адгезія лейкоцитів - білокрівці повільно котяться по поверхні ендотелію аж до повного зупинення, яке настає внаслідок контакту з ендотеліальними клітинами. В основі лежить взаємодія селектинів ендотелію з олігосахаридами, що входять до складу глікопротеїнів мембран лейкоцитів.
*Необоротна адгезія завершує процес крайового стояння лейкоцитів. Останні міцно зв'язуються з ендотелієм і більше не відриваються від нього. З часом уся поверхня ендотелію стає виставленою лейкоцитами, а отже нерівної такою ,що нагадує бруківку. Основу необоротної адгезії складає взаємодія лейкоцитарних інтегринів з подібними до імуноглобулінів молекулами мембран ендотеліальних клітин
5) агрегація еритроцитів - зменшення негативного заряду поверхні червонокрівців .
6) набрякання ендотеліальних клітин- веде до зменшення просвіту мікросудин
II. Позасудинні фактори:
1) здавлювання венозних судин набряковою рідиною;
2) втрата венулами еластичних властивостей внаслідок розщеплення колагену і еластину лізосомними ферментами (В. В. Воронін)
4. Стаз - припиненні течії крові в судинах передує явище передстазу для якого характерні поштовхоподібний( у систолу вперед, у діастолі на місці) або маятникоподібний(у систолу вперед, у діастолу назад) характер руху крові . За патогенезом стаз у вогнищі запалення має риси як венозного(внаслідок венозної гіперемії )і справжнього (капілярного) стазу. У розвитку останнього велику роль відіграють згущення крові( збільшення її в'язкості ) та агрегація еритроцитів.
70. Ексудація в осередку запалення, її причини і механізми. Фази підвищення проникності судинної стінки. Види ексудатів. Особливості геморагічного (на прикладі кору) та гнійного (на прикладі дифтерії) запалення у дитячому віці.
Ексудація — це вихід рідини і розчинених в ній компонентів плазми крові з кровоносних судин в тканину.
У основі ексудації лежать наступні механізми:
1) підвищення проникності судинної стінки;
2) збільшення гидростатічесого тиску в судинах;
3) збільшення осмотичного і онкотичного тиску в тканині.
Причини:
1) Власне судинні - підвищення проникностістінки судин.
2) Внутрішньосудинні – підвищення гідростатичного тиску крові у мікросудинах.
3) Позасудинні – зюільшення осмотичного і онкотичного тиску в осередку запалення.
Фази підвищення проникності судинної стінки :
1. Активація мікровезикулярного транспорту через ендотеліальні клітини.
2. Утворення наскрізних трансклітшних каналів в ендотеліоцитах (є наслідком значного посилення мікровезикулярного транспорту).
3. Збільшення просвіту міжендотеліальних щілин (відбувається в результаті скорочення й округлення ендотеліоцитів).
4. Десквамація (злущування) ендотелію. Є проявом первинної й вторинної альтерації.
5. Деполімеризація речовин, що з'єднують ендотеліальні клітини і є компонентами базальної мембрани судинної стінки.
6. Новоутворення судин – ангіогенез. У місцях відпупковування нових судин проникність клітинної стінки є дуже високою, поки не завершиться дозрівання ендотеліоцитів та утворення контактів між ними.
Види ексудатів:
1.Серозний ексудат складається переважно з води й альбумінів. Утворюється на початку багатьох видів запалення. Виявляється при алергічних запаленнях шкіри, опіках, механічних ушкодженнях, при запаленні слизових оболонок і при запаленні серозних порожнин (серозний плеврит, перикардит, перитоніт, артрит).
2. Катаральний (слизуватий) ексудат виникає при запаленні слизових оболонок носоглотки, легень, шлунково-кишкового тракту. Катаральні ексудати містять мукополісахариди, секреторні антитіла, лізоцим.
3. Фібринозний ексудат утворюється при сильних ушкодженнях ендотелію, що супроводжуються значним витоком високомолекулярного фібриногену. Фібриноген, що вийшов із судин, полімеризується в нитці фібрину і є характерним для деяких бактеріальних інфекцій - дифтерії, дизентерії. Виявляється при запаленні верхніх дихальних шляхів, товстого кишечника, перикарда, очеревини.
4. Гнійний ексудат містить гній, що складається з великої кількості життєздатних і зруйнованих нейтрофілів, уламків некротизованих тканин, частково розчинених шляхом ферментативного перетравлення. Утворюється найчастіше при інфекціях, викликаних піогенними бактеріями: стафілококами, стрептококами, пневмококами та ін.
5. Геморагічний ексудат містить значні кількості еритроцитів. Утворюється при важких ушкодженнях судин, що супроводжуються загибеллю ендотеліальних клітин і руйнуванням базальної мембрани. Виявляється при гострій грипозній пневмонії, сибірській виразці, отруєнні фосфогеном.
6. Гнилісний ексудат відрізняється наявністю продуктів гнильного розкладання тканин, внаслідок чого має брудно-зелене забарвлення і неприємний запах. Утворюється у випадку приєднання патогенних анаеробів.
71. Еміграція лейкоцитів в осередку запалення. Послідовність, причини і механізми еміграції лейкоцитів. Роль лейкоцитів у розвитку місцевих та загальних проявів запалення.
Еміграції лейкоцитів складається з 4 послідовних етапів: 1) крайове стояння; 2) проходження крізь судинну стінку; 3) міграція у тканини(хемотаксис); 4) здійснення функцій у осередку запалення. У еміграції лейкоцитів у вогнище запалення спостерігається певна черговість: спочатку емігрують нейтрофільні гранулоцити, потім — моноцити і, нарешті, — лімфоцити. Емігруючи в тканину, лейкоцити долають два бар'єри капілярної стінки: ендотелій і базальну мембрану. Нейтрофіли і макрофаги проходять через ендотелій по міжендотеліальних щілинах. Вони випускають свої псевдоподії в простори між ендотеліоцитами і "розсовують" клітини. Подолання базальної мембрани може бути обумовлене двома механізмами. Перший з них полягає в явищі тиксотропії — при контакті нейтрофіла з базальною мембраною її колоїди переходять із стану гелю в стан золя (відбувається розрідження мембрани). Нейтрофіл легко проходить через золь, після чого золь назад перетворюється на щільний гель. Другий механізм полягає у виділенні нейтрофілами нейтральних протеаз (еластаза, коллагенази), які розщеплюють волокнисті компоненти початкової мембрани.
Роль лейкоцитів у патогенезі запалення:
I. Стадія альтерації: Лейкоцити виробляють лейкотрієни, що спричинюють хемотаксис, скорочення гладких м’язів, набряк.
II. Стадія ексудації: Вихід лейкоцитів з просвіту судин через судинну стінку в навколишню тканину. При проходженні через базальну мембрану поліморфно-ядерний лейкоцит атакує її своїми ферментами (еластаза, коллагеназа, гіалуронідаза). Вони впливають на молекулярну структуру базальної мембрани, збільшуючи її проникність. У вогнищі запалення здійснюється активний рух лейкоцитів до хімічних подразників, якими можуть бути продукти протеолізу тканин. У вогнищі запалення головна функція лейкоцитів полягає в тому, щоб поглинати і переварювати чужорідні частки (фагоцитоз).
III. Стадія проліферації: Клітини перестають виробляти одні медіатори і починають синтезувати інші. Для завершення процесу запалення, особливо імунного, велике значення має зниження активності лімфоцитів. Гістамін через рецептори Н2 гальмує секрецію лімфокінів, знижує мітотичну активність лімфобластів, обмежує активність Т-кілерів. Лімфокіни інактівіруются також і макрофагами через один з монокінів гістаміну.
72. Фагоцитоз. Стадії та механізми. Роль фагоцитозу при запаленні. Порушення фагоцитозу та їх прояви у дітей.
Фагоцитоз — це активне поглинання клітинами твердого матеріалу. Клітини, що мають здатність здійснювати фагоцитоз, отримали назву фагоцитів. Розрізняють поліморфноядерні
(нейтрофіли) і мононуклеарнї фагоцити.
До мононуклеарних відносять облігатні фагоцити, які становлять систему мо-
ионуклеарних фагоцитів. У цю систему входять моноцити і клітини, що є їхніми
похідними: макрофаги сполучної тканини, клітини Купфера в печінці, альвеоля рні
макрофаги легень, макрофаги червоного кісткового мозку, вільні й фіксовані макрофаги
селезінки, макрофаги серозних порожнин, остеокласти, мікрогліальні клітини центральної
нервової системи.
Стадії:
Стадія зближення (хемотаксис):
Хемотаксис фагоцитів виникає під дією речовин, що отримали назву хемотак-cunie.
Розрізняють екзогенні й ендогенні хемотаксини. Екзогенними є бактеріальні
ліпополісахариди (ендотоксини), продукти руйнування бактеріальних стінок (мурамілдипептид). Ендогенними називають хемотаксини, що утворюються в самому організмі. Серед них найбільше значення мають побічні продукти активації комплементу (СЗа, С5а), лейкотрієни, лімфокіни і монокіни, фактор еміграції нейтрофілів.
Стадія прилипання:
Нерецепторними механізмами є електростатична й гідрофобна взаємодія поверхні фагоцита з об'єктом фагоцитозу. Оскільки поверхневий заряд фагоцитів негативний, то добре прилипають позитивно заряджені частки. Так само добре прилипають і частки з гідрофобними поверхнями.
Рецепторопосередковані механізми обумовлені існуванням на поверхні фагоцитів спеціальних рецепторів до речовин-опсонінів. Взаємодія фагоцита з об'єктом фагоцитозу відбувається через опсоніни (речовини, які сприяють прилипанню – IgG, C3b, С-реактивний білок), пов'язані з рецепторами.
3. Стадія поглинання:
1) Інвагінація плазматичної мембрани фагоцита в місці його контакту з об'єктом фагоцитозу.
2) Утворення фагосоми, що має свою мембрану і містить усередині об'єкт фагоцитозу.
3) Злиття фагосоми з лізосомами, у результаті чого утворюється фаголізосома.
Стадія перетравлювання:
1) Знищення (вбивання) бактерій - внутрішньоклітинний цитоліз. Здійснюється за допомогою бактерицидних систем фагоцитів.
2) Власне перетравлювання - гідроліз компонентів убитих бактерій за допомогою гідролітичних ферментів лізосом. Продукти, що утворилися при цьому, можуть бути використані фагоцитами для власних потреб.
Бактерицидними системами й речовинами фагоцитів є:
1) мієлопероксидазна система;
2) лізоцим;
3) лактоферин;
4) неферментні катіонні білки;
5) молочна кислота.
Роль у запаленні:
Макрофаги мають більшу тривалість життя у осередку запалення (ніж нейтрофіли), здатність розпізнавати, а потім поглинати ушкоджені нежиттєздатні клітини власного організму, в тому числі й нейтрофіли. З цим пов’язана їхня надзвичайна роль у «збиранні» запального ексудату.
Макрофаги – головні клітини, що беруть участь у розчиненні і видаленні з осередку запалення ушкодженої сполучної тканини, що необхідно для наступної реконструкції тканин. Вони синтезують і секретують нейтральні протеази, що руйнують зовнішньоклітинні колагенові й еластинові волокна сполучної тканини: еластазу, колагеназу, активатор плазміногену. Ферменти утворюються в результаті позаклітинного протеолізу сполучнотканинного матриксу, поглинаються потім макрофагами за допомогою спеціальних рецепторів і розкладаються внутрішньоклітинно із залученням лізосомальної системи.
Порушення фагоцитозу можливе при спадковій патології фагоцитів (порушенні метаболізму полісахаридів у лейкоцитах (хвороба Альдера), порушенні дозрівання фагоцитів (хвороба Чедіака-Хігаші) та ін., а також у результаті набутої недостатності фагоцитозу (променева хвороба, білкове голодування, старечий вік, тривала гормональна терапія та ін.).
Порушення фагоцитозу у дітей може проявлятись недостатністю імунної відповіді – первинні імунодефіцити (інфантильний летальний агранулоцитоз – синдроми Костманна, Швахманна), також порушення бактерицидного ефекту фагоцитів спостеріг. при хронічному гранулематозі.
73. Проліферація клітин в осередку запалення, її стадії та механізми. Механізми мітогенної дії факторів росту і цитокінів. Регенерація та фіброплазія як способи загоєння. Особливості проліферативного запалення у дитячому віці (на прикладі утворення туберкульоми).
Стадія проліферації включає:
1) розмноження клітин, тобто власне проліферацію;
2) синтез позаклітинних компонентів сполучної тканини — колагену, еластину, протеогліканов, глікопротеїн-1.
Механізми:
- інгібування ферментів (інгібітор – α2-макроглобулін);
- припинення руйнівних явищ (усунення вільних радикалів);
- пригнічення активності клітин запалення;
- зменшення активності лімфоцитів;
- ендокринні фактори (кортикостерон та гідрокортизон – спричинюють лімфопенію)
Фактори росту:
I. Зменшення концентрації в тканині кейлонів. Кейлони-це речовина білкової природи, які утворюються зрілими клітинами. Вони є інгібіторами клітинного ділення. При пошкодженні і загибелі клітин у вогнищі запалення концентрація кейлонів зменшується, а отже, знімається гальмівний вплив на малодиференційовані (камбіальні) клітки. Вони починають ділитися. Ділення продовжується до тих пір, поки концентрація кейлонів не збільшиться до рівня, який існував в неушкодженій тканині.
II. Збільшення концентрації в тканині стимуляторів проліферації — чинників зростання. Чинники зростання поступають в тканину з плазми крові або є продуктами клітин, що знаходяться у вогнищі запалення. Їх дія вказаних регулювальників здійснюється через активацію внутрішньоклітинних протеїнкіназ.
Цитокіни - це збірне поняття для позначення великої групи біологічно активних речовин білково-пептидної природи, що регулюють взаємодію між різними типами клітин (4 групи: інтерлейкіни, інтерферони, гемопоетичні колонієстимулятивні фактори, фактори що пригнічують ріст пухлин). Однією з властивостей багатьох цитокінів є їхня мітогенна активність, що виявляє себе посиленням процесів проліферації в осередку запалення. Водночас, деякі цитокіни (ІЛ-1, ФНП) стимулюють синтез колагену і новоутворення кровоносних судин (ангіогенез).
Регенерація – це проліферація (розмноження) попередників паренхіматозних клітин, що залишаються. У цьому випадку відбувається заміна втрачених клітин, клітинами того самого типу. Регенерація характерна для тканин, клітини яких постійно проліферують (епітеліальна і кровотворна) і тканин, що в нормі мають низький рівень реплікації, але за необхідності, під впливом відповідних факторів росту можуть розпочати компенсаторну гіперплазію (глвдком’язеві клітини та паренхіматозні клітини печінки, нирок і підшлункової залози).
Фіброплазія – відновлення і новоутворення сполучної тканини/розмноження мезенхімальних клітин строми, з подальшим диференціюванням їх у фібробласти, продукцією фібробластами колагену та глікозаміногліканів, з яких формуються фібрилярні структури і грануляційна тканина, що перетворюється на сполучну тканину. Здійснюється за рахунок активації проліферації фібробластів, епітеліоцитів, гладком’язевих клітин під впливом ЕФР, ФРТ, ФРФ,ТФР-α, СЕФР тощо. Вона може відбутися навіть при повній загибелі тканини у разі потрапляння в ушкоджену тканину стромальних стовбурових клітин червоного кісткового мозку (вони мультипотентні). Під впливом ТФР-β вони перетворюються на фібробласти, адипоцити, хондроцити, остеобласти, міобласти і ендотеліоцити.
Туберкульома відноситься до специфічних гранульом. Специфічне запалення за характером клітинних реакцій та наявності вогнищ казеозного некрозу має специфічні для їх збудника морфологічні особливості. Туберкульозна гранульома має чітку зональність: у її центрі розташоване вогнище некрозу, по периферії – вал з епітеліоїдних клітин, лімфоцитів, макрофагів, плазматичних клітин і гігантських клітин Пирогова-Лангханса., мало судин. При забарвленні за Цілем-Нільсеном у гігантських клітинах виявляються мікобактерії туберкульозу.
Механізм утворення гранульоми складається з 4-х стадій:
Нагромадження у вогнищі ушкодження юних моноцитів.
Дозрівання цих клітин у макрофаги й утворення макрофагальної гранульоми.
Трансформація макрофагів в епітеліоїдні клітини й утворення епітеліоїдно-клітинної гранульоми.
Злиття епітеліоїдних клітин (або макрофагів) з утворенням гігантських клітин (клітин сторонніх предметів або клітин Пирогова-Лангханса). Гігантські клітини можуть містити до 100 і більше ядер.
74. Роль реактивності організму в розвитку запалення. Зв’язок між паталогічною імунною відповіддю і запаленням. Вплив гормональних чинників на запалення.
Перебіг запальної реакції залежить від реактивності організму, що визначається станом нервової, ендокринної та імунної систем. Еволюційне формування усіх цих систем, а також судинної, сприяло ускладненню і удосконаленню запалення, як захисної реакції на пошкодження тканини.
Стан нервової системи визначає ступінь вираженості первинного пошкодження, а також можливості виникнення запалення. Значення цієї системи в динаміці розвитку патологічного процесу підтверджується чисельними випадками виникнення ознак опіку у пацієнтів під впливом гіпнозу. В практиці психіатричної клініки неодноразово виникали випадки розвитку гіперергічного запалення при місцевій дії пошкоджуючого агента за умов маніакального збудження, а під час важкої депресії запальна реакція протікала дуже в’яло.
Зміни нервово-імпульсних та нервово-трофічних впливів на пошкоджену тканину можуть сприяти підсиленню мікроциркуляції. Нейромедіатори і речовини-трофогени, які виділяються при пошкодженні нервових закінчень у вогнищі запалення, здатні впливати на активність фагоцитів, стимулюючи в них вільнорадикальні процеси. Нервовотрофічні впливи визначають ступінь зрілості тканини, її здатність до відновлення білкового складу, біогенезу ультраструктур, проліферацію клітин і міжклітинної речовини. Порушення аферентної інервації підсилює альтеративні процеси і сповільнює репарацію паренхіматозних клітин.
. Активну участь в регуляції проліферативно-відновних процесів приймають нейропептиди, особливо опіоїдні пептиди. Стимуляція цими пептидами опіоїдних рецепторів С-волокон зменшує відчуття болю, пригнічує виділення норадреналіну з симпатичних нервових закінчень, припиняє активацію лаброцитів і тромбоцитів, усуває розлади мікроциркуляції, порушення гемостазу, зменшує активність вільнорадикальних процесів, які залежать від катехоламінів. Запалення в процесі філогенезу сформувалося як захисна реакція організму теплокровних особин. Організм захищається від пошкоджуючого агента завдяки відмежуванню пошкодженої ділянки. Навколо вогнища запалення утворюється бар’єр, який пропускає різні речовини лише в одному напрямку (в центр вогнища) завдяки закупорці вивідних кровоносних та лімфатичних судин. У вогнищі запалення створюються неблагоприємні умови для розвитку мікроорганізмів. Але значне пошкодження тканини чи порушення мікроциркуляції може провокувати суттєві розлади метаболізму, значну гіпоксію, загальну інтоксикацію. Запалення є яскравим прикладом патологічного процесу, який відображає одночасне поєднання елементів пошкодження (полому) та протидії організму (міра проти полому), що спрямоване на виживання. "Білки гострої фази запалення" ("реактанти гострої фази") - це білки, концентрація яких у плазмі крові при гострому запаленні збільшується більш як на 50 %. Вони утворюються в основному в печінці під впливом: а)продуктів, що надходять у кров з вогнища запалення (продуктів альтерації); б)інтерлейкіну-1 макрофагального походження. Нині відомо більше 10 "білків гострої фази запалення". Всі вони в основному мають захисне значення. До них відносять:. 1)інгібітори протеаз (орозомукоїд, аантитрипсин); 2)антиоксиданти (гаптоглобін, церулоплазмін); 3)імуноглобуліни й антитілоподібніречовини (антитіла, С-реактивний білок).
Запалення, як і будь-який патологічний процес, являє собою єдність і боротьбу двох начал: власне патологічного і захисно-компенсаторного. У запаленні завжди наявні руйнівний і захисний компоненти. Власне патологічними явищами при запаленні слід вважати первинну і вторинну альтерацію, набряк, фізично-хімічні зміни й порушення обміну речовин у вогнищі запалення, біль і порушення функції. Захисне значення мають еміграція лейкоцитів, знищення флогогенних агентів, активація процесів репарації ушкодженої тканини, загальні реакції організму, спрямовані на збільшення його резистентності (гарячка, лейкоцитоз, збільшення вмісту "білків гострої фази запалення").
За механізмом впливу на запалення гормони поділяють на прозапальні й протизапальні. Прикладом прозапальних гормонів, тобто гормонів, що посилюють запалення, можуть бути мінералокортикоїди зокрема альдостерон- Під його впливом істотно підвищується проникність стінок судин, у результаті чого збільшується ексудація. Найважливішими протизапальними гормонами є глюкокортикоїди. У високих (лікувальних) дозах вони викликають дерепресію гена, що кодує структуру особливого білка - ліпокортину. Ліпокортин є природним внутрішньоклітинним інгібітором фосфоліпази А2.
Пригнічення цього ферменту ліпокортином має два важливих наслідки:
1)з одного боку, зменшується утворення лізофосфоліпідів, у результаті чого зменшуються явища альтерації (стабілізація мембрани);
2)з другого боку, зменшується утворення арахідонової кислоти та її похідних (простагландинів, тромбоксанів, простациклінів, лейкотрієнів). Ця обставина пояснює інші протизапальні ефекти глюкокортикоїдів — зменшення ексудації і еміграції лейкоцитів, ослаблення порушень мікроциркуляції. У результаті пригнічення процесів клітинного поділу й біосинтезу білка зменшуються явища проліферації у вогнищі запалення.
75. Гарячка: визначення поняття, принципи класифікації. Зв’язок між гарячкою і запаленням. Види пірогенів. Утворення пірогенів при інфекції , асептичному ушкодженні та імунних реакціях. Хімічна природа і походження вторинних пірогенів, механізм їх дії.
Гарячка - це типовий патологічний процес, що розвивається у вищих гомойотермних організмів у відповідь на пірогенні подразники і виявляє себе перебудовою терморегуляції, спрямованою на активне підвищення температури тіла.
Постійна, або стійка гарячка. Спостерігається постійно підвищена температура тіла і протягом доби різниця між ранковою і вечірньою температурою не перевищує 1°С. Вважається, що подібне підвищення
температури тіла характерне для крупозного запалення легенів, черевного тифу, вірусних інфекцій (наприклад, грипу).
Послаблювальна гарячка (ремітуюча). Спостерігається постійно підвищена температура тіла, але добові коливання температури перевищують 1°С. Подібне підвищення температури тіла зустрічається при туберкульозі, гнійних захворюваннях (наприклад, при тазовому абсцесі, емпіємі жовчного міхура, рановій інфекції), а також при злоякісних новоутвореннях.
Переміжна гарячка (інтермітуюча). Добові коливання перевищують 1 °С, але тут ранковий мінімум лежить в межах норми. В цьому випадку підвищена температура тіла з'являється періодично, приблизно через рівні проміжки (найчастіше біля полудня або вночі) на декілька годин. Переміжна гарячка особливо характерна для малярії, а також її спостерігають при генерализованій цитомегаловірусній інфекції, гнійній інфекції (наприклад, холангіті).
Виснажлива гарячка (гектична). Вранці, як і при інтермітуючій, спостерігають нормальну або навіть знижену температура тіла, але ось добові коливання температури доходять до 3-5°С і часто супроводжуються масивним потовиділенням, що виснажує. Подібне підвищення температури тіла характерне для активного туберкульозу легенів і для септичних захворювань.
Зворотна, або збочена гарячка відрізняється тим, що ранкова температура тіла більша за вечірню, хоча періодично все одно буває звичайне невелике вечірнє підвищення температури. Зворотна гарячка зустрічається при туберкульозі (частіше), сепсисі, бруцельозі.
відношення до інфекції, наприклад, при переливанні крові, при введенні білків і ліпідів з метою парентерального харчування. Неінфекційна гарячка поділяється на два види - гарячка при асептичному запаленні і гарячка при алергії. Патогенез їх однаковий - вони викликаються інтерлейкіном 1. Пусковим механізмом утворення і виділення інтерлейкіну 1 також вважається фагоцитоз: при асептичних запаленнях (інфаркт міокарда, хвороби сполучної тканини, запальні реакції у відповідь на пухлини) - фагоцитоз загиблих і пошкоджених клітин або клітинних фрагментів; при алергіях - фагоцитоз комплексів антиген-антитіло.
Інтерлейкін 1 - білок низької молекулярної маси, який викидається фагоцитуючими клітинами в кровоток і досягає центра терморегуляції. Це дуже могутній стимулятор центра, він викликає гарячку в нанограмових кількостях. У фагоцитуючих клітинах його нема в готовому вигляді, він утворюється в процесі фагоцитозу. Секреція інтерлейкіну 1 не супроводжується загибеллю фагоцитів.
Центр терморегуляції, на який діє інтерлейкін 1, локалізований в передній частині гіпоталамуса біля дна третього шлуночка. Пірогенними речовинами називаються такі речовини, що, потрапляючи в організм ззовні або утворившись усередині нього, викликають гарячку. По походженню пірогенні речовини розділяються на екзогенні (бактеріальні, небактеріальні) і ендогенні (лейкоцитарні), а по механізму дії ‑ на первинні і вторинні. Мікроорганізми містять у собі пірогенні речовини (зовнішні, екзогенні пірогени). Вони є складовою частиною мікробних токсинів. Найбільше вивчені в цьому плані ендотоксини грамнегативних бактерій. Вони складаються з трьох фракцій - ліпідної, полісахаридної і білкової. Пірогенні властивості має ліпідна фракція (ліпоїд А). Із грамнегативних бактерій вдалося виділити високоочищені пірогени і створити препарати для клінічного застосування - пірогенал, пірексаль, піромен. Діючою речовиною у них є ліпоїд А. Вихідним матеріалом для одержання пірогенів як лікарських засобів послужили грамнегативні бактерії. Вітчизняний препарат пірогенал отриманий з Pseudomonas aerugenosa, швейцарський пірексаль був виділений з Salmonella abortus equi. Первинні пірогени можуть утворюватися в самому організмі, незалежно від мікроорганізмів (гарячка при переломі кісток, при переливанні крові, при інфаркті міокарда). Ці "жаронесучі" речовини утворюються при ушкодженні або руйнуванні власних тканин і здійснюють на організм дію, аналогічна дії "справжніх", тобто мікробних пірогенів.
Вторинними називають лейкоцитарні пірогени, які утворюються і вивільнюються лейкоцитами під впливом первинних пірогенів. Вторинні пірогени мають здатність безпосередньо впливати на центр терморегуляції й викликати розвиток гарячки. Сьогодні відомо, що одним із вторинних лейкоцитарних пірогенів є інтерлейкін-1. Ця речовина утворюється і вивільнюється лейкоцитами (нейтрофілами й макрофагами) у процесі активації фагоцитозу. Поряд з дією на центр терморегуляції інтерлейкін-1 виявляє цілу низку інших ефектів, що визначають клінічні прояви гарячки.
76. Гарячка: стадії розвитку, зміни терморегуляції, обміну речовин та фізіологічних функцій. Захисне значення та патологічні прояви гарячки. Принципи жарознижувальної терапії. Поняття про піротерапію.
Гарячка — це захисно-пристосовна реакція організму, що виникає у відповідь на дію патогенних подразників і характеризується перебудовою процесів терморегуляції, що призводять до підвищення температури тіла і стимулює природну реактивність організму. Біологічне значення гарячки полягає в активізації імунологічного захисту
Виділяють три стадії гарячки :
1.Підвищення (st. incrementi),
2.Високогостояння (st. fastigi)
3.Зниженнятемператури (st. decrementi).
Упершійстадіїтемпературатілапідвищується, удругій— утримуєтьсяякийсьчаснапідвищеномурівні, утретій —знижуєтьсядовихідногорівня.
Стадіяпідйомутемператури - Stadiumincrementi.
В цю стадію процес теплоутворення переважає над тепловіддачею. Периферічні судини шкіри звужені, потовиділення зижене. В цю стадію нерідко у тварин спостерігається «озноб» - відчуття холоду, що пояснюється звуженням судин шкіри, дією на температурні рецептори шкіри, а також зниженням порогу чутливості центру терморегуляції до холодового подразнення.
Стадія стояння високої температури - Stadium factigii. Характеризується тим, що висока температура тіла з певними добовими коливаннями утримується. В цю стадію спостерігається посилене утворення тепла в організмі і збільшення тепловіддачі – підтримується відносний нестійкий баланс між посиленим теплоутворенням і посиленою тепловіддаче
Стадія зниження температури – Stadium decrementi. В цю стадію процес тепловіддачі посилюється і температура тіла знижується.
Зниження температури тіла може бути повільним – лізис, або швидким – криза.
Захисно-пристосувальне значення гарячки: При гарячці в організмі створюються несприятливі умови для розвитку збудників інфекційних хвороб і підвищується активність механізмів неспецифічної і специфічної резистентності організму.
Зокрема: -пригнічується розмноження багатьох вірусів, посилюється утворення інтерферонів.
-збільшується фагоцитарна активність макрофагів і нейтрофілів.
-підвищується інтенсивність синтезу антитіл.
-зростає чутливість багатьох інфекційних збудників до дії ЛЗ.
Власне патологічні прояві гарячки: залежать від рівня температури тіла( що вища температура тіла,тим вищі негативні прояви):
-гарячка завдає страждань хворому: порушується загальний стан хворого, стають характерним нездужання, головний біль, відчуття ознобу, жар тощо.
-значних змін зазнає обмін речовин в організмі. Збільшується основний обмін ( на 10-12%) це викликає збільшення споживання кисню і поживних речовин.
- зростає потреба у воді - розвівається зневоднення.
- гіпокапнія, газвовий алкалоз.
Принципи жарознижувальної терапії: основним патогенетичним принципом є зниження “установної точки” центру терморегуляції, що досягається пригніченням утворення простагландину Е2, за допомогою інгібіторів циклооксигенази або інгібіторів фосфоліпазі А2.
Піротерапія – піротерапією називають метод лікування за допомогою штучного підвищення температури тіла. У даний час піротерапію застосовують з метою прискорення репаративних процесів для усунення наслідків травматичних і запальних процесів у ЦНС, травматичних пошкоджень спинного мозку і периферичних нервів, опіків очей, для розсмоктування рубців, спайок, із метою підвищення проникності ГЕБ для поліпшення доступу лікарських препаратів, антитіл в головний мозок, що має особливо важливе значення при лікуванні сифілісу на пізніх стадіях хвороби.
77. Особливості розвитку запалення і гарячки у дитячому віці. Патогенетичне обґрунтування застосування протизапальної та жарознижувальної терапії у дитячому віці.
(див питання 76)
Запалення - це типовий патологічний процес, який виникає у відповідь на пошкодження тканин і складається зтрьох взаємопов’язаних судинно-тканинних компонентів: альтерація, порушення мікроциркуляції зексудацією та еміграцією лейкоцитів, проліферація.
Стадії. Запалення складається з трьох стадій (перша – альтерація; друга - порушення мікроциркуляції з ексудацією та еміграцією лейкоцитів увогнищі запалення, третя - проліферації.
Альтерація. Перша стадія запалення альтерація з якої починаються всі форми запалення, характеризується пошкодженням, тобто порушенням структури і функції клітин, волокнистихструктур, мікроциркуляторного русла, основної речовини, нервових рецепторів та волокон. Структурні зміни в тканині викликають порушення метаболізму білків, жирів, вуглеводів, фізико-хімічні зміни, найважчим наслідком яких є некробіоз інекроз. Розрізняють первинну та вторинну альтерацію.
Порушення мікроциркуляції у вогнищі запалення. Запалення характеризується місцевими порушеннями циркуляції крові і лімфи, особливо в мікроциркуляторному руслі (артеріоли, метаартеріоли, капіляри, венули).
5 стадій порушення мікроциркуляції, що спостерігаються у вогнищі запалення:
І стадія – короткочасний спазм судин;
ІІ стадія - артеріальна гіперемія;
ІІІ стадія - венозна гіперемія;
ІV стадія – передстаз;
V стадія - стаз.
Ексудативні процеси у вогнищі запалення (ексудація). Реакція мікроциркуляторного русла, що виникає одночасно з пошкодженням, є важливим компонентом запалення і характеризується фізико-хімічними змінами судинної стінки. Головною причиною судинної реакції та послідовного збільшення проникності судин є дія медіаторів запалення, таких як гістамін, серотонін, кініни, простагландини, клітинні ферменти та ін. Збільшення проникності сприяє розвитку ексудації та еміграціїлейкоцитів (переміщення лейкоцитів з крові у вогнище запалення).
Еміграція лейкоцитів. Судинні зміни у вогнищі запалення та сповільнення кровотоку викликають перерозподіл форменних елементів. Лейкоцити виходять за межі осьового циліндра та плазматичного шару, підходять до стінки судини і починають прикріплятися до неї. Виникає фаза крайового стояння лейкоцитів. Подальше сповільнення течії крові сприяє активному прилипанню лейкоцитів (адгезії), які утворюють муфту вздовж стінки судини, порушуючи ламінарність руху рідини.
Проліферація. Даний процес принципово не залежить від типу пошкоджуючого фактора.
Процес проліферації у вогнищі запалення регулюється речовинами, які можуть стимулюваи (мітогени) чи пригнічувати (кейлони) розмноження клітин. Тканинними джерелами регенераційного матеріалу є камбіальні клітини – родоначальники усіх інших диференційованих клітинних компонентів конкретної тканини.
78. Основні відмінності між гарячкою, екзогенним перегріванням та іншими видами гіпертермії. Експериментальні моделі гарячки, екзо- та ендогенної гіпертермії.
Відмінності між гарячкою та гіпертермією: Гіпертермія розвивається внаслідок зовнішнього або внутрішнього розігрівання організму. При гарячці центр терморегуляції перебудовується таким чином, що це дозволяє активно піднести температуру тіла на вищий рівень. При гіпертермії температура тіла підвищується всупереч спробам центру терморегуляції утримати її на нормальному рівні.
79. Артеріальна і венозна гіперемії: визначення поняття, прояви, види, причини і механізми розвитку, варіанти завершення і наслідки. Дослід К. Бернара.
Артеріальна гіперемія - це збільшення кровонаповнення органа або тканини за рахунок надмірного надходження крові по артеріальних судинах.
При артеріальній гіперемії спостерігають розширення дрібних артерій, артеріол, вен і капілярів; прискорення течії крові в них, пульсацію дрібних артерій і капілярів, збільшення числа видимих оком судин, збільшення тиску в артеріолах, капілярах і венах. У результаті зазначених змін виникає почервоніння, підвищується місцева температура, збільшується об'єм гіперемованої ділянки, підвищується тургор тканини, посилюються обмін речовин і функція органа.
Причиною артеріальної гіперемії може бути вплив фізичних, хімічних і біологічних факторів зовнішнього середовища; збільшення навантаження на орган або ділянку тканини; психогенні впливи.
Фізіологічною називають артеріальну гіперемію, що виникає під дією звичайних фізіологічних подразників (збільшення навантаження на орган, психогенні впливи). Основними її різновидами є робоча й реактивна гіперемія.
Робоча гіперемія - це збільшення течії крові в органі під час посилення його функції (збільшення вінцевого кровообігу при посиленні роботи серця, гіперемія слинних залоз під час приймання їжі та ін.).
Реактивна гіперемія являє собою збільшення течії крові після його короткочасного обмеження. Розвивається зазвичай у нирках, головному мозку, шкірі, кишках, м'язах.
Патологічна артеріальна гіперемія виникає під дією незвичайних (патологічних) подразників або в результаті підвищення чутливості судин до звичайних впливів. Вона супроводжує розвиток таких патологічних процесів, як запалення, алергія, опіки, гарячка, її клінічними прикладами можуть бути інфекційний або алергічний висип, почервоніння обличчя при багатьох інфекційних хворобах (кір, скарлатина, висипний тиф), почервоніння половини обличчя при невралгії трійчастого нерва і т. д.
Розрізняють два механізми: нейрогенний і пов'язаний з дією місцевих хімічних (метаболічних) факторів.
Нейрогенна артеріальна гіперемія залежно від конкретних механізмів її розвитку може бути нейротонічного і нейропаралітичного типів.
Гіперемію, пов'язану з дією місцевих гуморальних факторів, іноді називаютьмі-
опаралітичною, підкреслюючи тим самим первинність порушень скоротливих властивостей гладких м'язів судин під впливом хімічних агентів.
Нейротонічний тип артеріальної гіперемії розвивається при збільшенні імпульсації по судинорозширювальних нервах (вазодилататорах). До останніх належать парасимпатичні нерви і симпатичні холінергічні нерви, медіатором яких є ацетилхолін.
В експерименті на тваринах цей тип артеріальної гіперемії відтворюють шляхом подразнення судинорозширювальних нервів. Так, подразнення chorda tympani (гілка n. facialis) викликає артеріальну гіперемію і посилення секреції піднижньощелепної слинної залози (дослід К. Бернара).
У клініці нейротонічна гіперемія виникає найчастіше рефлекторно у зв'язку з подразненням екстеро- та інтерорецепторів, а також при подразненні судинорозширювальних нервів та їх центрів. Цим, зокрема, пояснюється почервоніння обличчя і шиї при патологічних процесах у внутрішніх органах (яєчниках, серці, печінці).
Нейропаралітична артеріальна гіперемія розвивається при припиненні імпульсації по симпатичних адренергічних нервах, що мають судинозвужувальну дію. В експерименті на тваринах її моделюють за допомогою хірургічних втручань і фармакологічних способів. Часто застосовують перетинання симпатичних адренергічних волокон і нервів. Так, при перетинанні симпатичних волокон сідничного нерва спостерігають розширення судин лапки жаби (дослід А. Вальтера), а при видаленні шийного вузла симпатичного стовбура-
почервоніння й підвищення температури вуха кроля з боку операційного втручання (дослід К. Бернара).
Наслідки
У більшості випадків артеріальна гіперемія супроводжується збільшенням інтенсивності обміну речовин і посиленням діяльності органа, що є пристосуванням до дії підвищеного функціонального навантаження.
Однак можливі й несприятливі наслідки. При артеріосклерозі, наприклад, різке розширення судини може супроводжуватися розривом її стінки і крововиливом у тканину. Особливо часто подібне явище спостерігається в головному мозку.
Венозна гіперемія - це збільшення кровонаповнення органа або ділянки тканини в результаті утрудненого відтоку крові по венах.
Порушення відтоку крові по венах може бути пов'язане з такими чинниками:
1) внутрішньосудинними (закупорка вен тромбом або емболом);
2) позасудинними (здавлювання вен пухлиною, рубцем, збільшеною маткою, набряковою рідиною);
3) факторами самої судинної стінки (конституціональна слабкість еластичного апарату вен, недостатній розвиток і знижений тонус гладком'язових елементів їхніх стінок);
4) порушеннями загальної гемодинаміки (ослаблення функції правого шлуночка серця, зменшення присмоктувальної дії грудної клітки, утруднення течії крові в малому колі кровообігу).
Для венозної гіперемії характерне збільшення об'єму органа або ділянки тканини, ціаноз, місцеве зниження температури, набряк, підвищення тиску у венах і капіля-
pax застійної ділянки, уповільнення течії крові, вихід еритроцитів за межі судинного русла (діапедез). На завершальному етапі гіперемії можливий маятникоподібний рух крові і стаз. Тривале розширення вен призводить до розтягнення їхньої стінки, що може супроводжуватися гіпертрофією м'язової оболонки, явищами флебосклерозу і варикозного розширення вен.
Місцеві зміни при венозній гіперемії пов'язані в основному з кисневим голодуванням (гіпоксією) тканини. Гіпоксія при цьому спочатку обумовлена обмеженням припливу артеріальної крові, а потім дією на тканинні ферментні системи продуктів порушеного обміну, наслідком чого є порушення утилізації кисню.
Кисневе голодування при венозній гіперемії обумовлює розлади тканинного метаболізму, викликає атрофічні і дистрофічні зміни, надмірне розростання сполучної тканини (наприклад, цироз печінки при венозному застої, викликаному недостатністю функції серця).
Якщо венозна гіперемія має генералізований характер, то можливим є ряд
загальних гемодинамічних порушень з важкими наслідками. Найчастіше вони виникають при закупорці великих венозних судин - ворітної, нижньої порожнистої вен. Скупчення крові в зазначених судинних резервуарах (до 90 % всієї крові) супроводжується різким зниженням артеріального тиску, порушенням живлення життєво важливих органів (серця, мозку), що може призвести до смерті.
80. Ішемія: визначення поняття, прояви, види, причини, механізми розвитку, наслідки. Механізми ішемічного пошкодження клітин. Синдром ішемія – реперфузія.
Ішемія - це зменшення кровонаповнення органа або ділянки тканини в результаті обмеження або повного припинення припливу артеріальної крові. Ішемію називають ще місцевим недокрівя
Ішемія характеризується зблідненням ділянки органа, зниженням його температури, порушенням чутливості (відчуття оніміння, поколювання, "повзання мурашок"), больовим синдромом, зменшенням швидкості течії крові і об'єму органа, зниженням артеріального тиску на ділянці артерії, розташовані нижче перешкоди; зниженням напруги кисню в ішемізований ділянці органа або тканини, зменшенням утворення тканинної рідини і зниженням тургору тканини, порушенням функції органа або тканини, дистрофічними змінами.
Основними типами ішемії є компресійна, обтураційна і ангіоспастична.
Компресійна ішемія виникає в результаті здавлювання артерій ззовні лігатурою, рубцем, пухлиною, стороннім предметом та ін.
Обтураційна ішемія є наслідком часткового звуження або повного закриття просвіту артерій атеросклеротичною бляшкою, тромбом або емболом.
Ангіоспастична ішемія виникає внаслідок спазму артерій, викликаного емоційним впливом (страх, хвилювання, гнів), фізичними факторами (холод, травма, механічне подразнення), хімічними агентами, біологічними подразниками (токсини бактерій) і т. д. В основі спазму можуть лежати нервові рефлекторні механізми або безпосередня дія подразників на гладкі м'язи судин (вплив вазопресину, ангіотензи-
ну II, ендотеліну).
Виділяють такі стадії патогенезу
I. Порушення енергетичного обміну. Вони виявляють себе зниженням ефективності циклу Кребса і тканинного дихання, активацією гліколізу, а в кінцевому підсумку - зменшенням вмісту в клітинах макроергічних сполук —
креатинфосфату і АТФ. Порушення утворення енергії на ділянці ішемії патогенетично пов'язане з недостатньою доставкою кисню й необхідних для окиснення субстратів, зниженням активності й синтезу ферментів, виходом ферментів з ушкоджених клітин, роз'єднанням окиснення й фосфорування.
II. Порушення енергозалежних процесів у клітинах, викликане зменшенням утворення енергії. При цьому порушуються специфічні функції клітин (скоротлива, секреторна та ін.), механізми активного транспорту речовин, зокрема, робота іонних насосів; знижується біосинтез білків неколагенового типу, що беруть участь у структурній організації клітин і тканин. В остаточному підсумку розвиваються ушкодження клітин і некробіотичні зміни, які в найважчих випадках закінчуються утворенням осередку некрозу - інфаркту.
III. Посилення біосинтезу компонентів сполучної тканини - колагену, глікозаміногліканів, глікопротеїнів, що є основою для наступного склерозування ішемізо-
ваної ділянки тканини або органа.
Патофізіологія фази ішемії
Патофізілогія синдрому ішемії-репер-фузії складається із сукупності патологічних процесів, що відбуваються в ішемічну і реперфузійних фази, які невід'ємно пов'язані між собою.
При раптовому порушенні артеріального кровообігу нижньої кінцівки настає гостра ішемія, в результаті чого виникає пошкодження тканин нижньої кінцівки. В основі механізму пошкодження лежить циркуляторная гіпоксія, що призводить до виснаження запасів кисню і високоенергетичних субстратів клітини, таких як аденозинтрифосфат (АТФ) і КРЕА-тінфосфат (КФ).
81. Тромбоз: визначення поняття, види тромбів. Причини, механізми, наслідки тромбоутворення.
Тромбоз – це процес прижиттєвого утворення
в просвіті кровоносних судин і в порожнинах серця згустків крові, що
складаються з її елементів.
Класифікація тромбів : а) за будовою : білі, червоні, змішані; б) за місцем
утворення : артеріальні, венозні; в) пристінкові, закупорювальні.
Причини: тріада Вірхова : ушкодження ендотелію судинної стінки + порушення
течії крові + підвищення здатності до зсідання.
Механізми: Внутрішній механізм тромбоутворення. Запускається
ушкодженням ендотелію (альтерацією), причиною якого може бути запалення,
алергія та атеросклероз. Ушкодження ендотелію призводить до оголення адгезивних
білків субендотелію (колаген, фібронектин, тромбоспондин, фактор Віллебранда)
та активації тромбоцитів. Адгезивні білки зв’язуються із глікопротеїновими
рецепторами (інтегринами) тромбоцитів і сприяють їхньому прикріпленню (адгезії)
до судинної стінки. При активації тромбоцитів виникає утворення і вбудовування
у мембрану цих клітин рецепторів агрегації та адгезії тромбоцитів. Пошкоджені
ендотеліоцити судин виділяють біологічно активні речовини (фактор агрегації
тромбоцитів (ФАТ), АДФ, тромбін), що активують тромбоцити.
Крім того, активація тромбоцитів проходить і під впливом катехоламінів та серотоніну, що з’єднуються із рецепторами клітин. Як наслідок підвищується рівень внутрішньоклітинного Са2+ за рахунок його виходу із внутрішньоклітинного депо (“електронно-щільних” гранул) та збільшується надходження Са2+ до тромбоцитів із позаклітинного середовища. Підвищення концентрації Са2+ в протоплазмі тромбоцитів сприяє дегрануляції a-гранул тромбоцитів (вивільнення тромбоспондину, фактору Віллебранда, фібриногену). Зазначені біологічно активні речовини забезпечують агрегацію і адгезію тромбоцитів. Із “електронно-щільних” гранул у позаклітинне середовище також надходять серотонін і АДФ. В свою чергу, іони кальцію активують фосфоліпазу А2, яка вивільняє з фосфоліпідів мембран тромбоцитів арахідонову кислоту і, тим самим, забезпечує утворення тромбоксану А2. Тромбоксан А2 також сприяє виходу іонів Са2+ з “електронно-щільних” гранул тромбоцитів. Активація тромбоцитів викликає збільшення кількості адгезивних рецепторів на їхній мембрані.
Білкові речовини – тромбоспондин, фактор
Віллебранда, фібриноген утворюють “містки” між рецепторами тромбоцитів і
рецепторами білків субендотеліального шару мікросудин.
Клітинна фаза завершується тим, що на ушкодженій ділянці судинної стінки
осідають і прикріплюються тромбоцити, тісно пов’язані між собою і субендотелієм
судин. Утворений
тромбоцитарний прошарок, білки субендотелію, а також кініноген плазми крові є
активаторами плазмового XII фактору згортання крові (фактору Хагемана). Так
запускається внутрішній механізм каскадної активації
плазмових факторів згортання крові.
Фактор Хагемана є активатором XI фактору (плазмового попередника тромбопластину), який у активному стані позначається як фактор XIa. Фактор XIa в присутності іонів Са2+ і фосфоліпідів активує фактор IX (активний Крістмас-фактор, IXa), який, утворюючи комплекс із VIII фактором (антигемофільним глобуліном А) і іонами Са2+, активує Х фактор (фактор Стюарта-Прауера). В свою чергу, активований Х фактор (фактор Ха) завдяки об’єднанню із V фактором (проакселерином), іонами Са2+ і фосфоліпідами утворює активний фермент, що перетворює фактор II (протромбін) у тромбін (фактор IIа).
Під дією тромбіну у присутності іонів Са2 від фібриногену (фактор I) відщеплюються чотири поліпептиди, і він перетворюється у фібрин-мономер, який у подальшому при полімеразації переходить у стан фібрину-полімеру. Ця форма фібрину без стабілізації може легко розщеплятися протеїназами крові. Стабілізація фібрину здійснюється активованим XIII плазмовим фактором згортання (фібринстабілізуючим фактором), який стає активним (тобто перетворюється у фактор XIIIa) під впливом тромбіну (фактора IIa) і іонів Са2+. В результаті згусток (тромб) стає міцним, еластичним і стійким до дії протеаз, а саме, плазміну. В нитках стабілізованого фібрину, зчеплених між собою і прикріплених до ушкодженої ділянки судини, розміщуються формені елементи крові – еритроцити і лейкоцити.
Через деякий час згусток крові (тромб) стискається (стадія “ретракції тромбу”), витискаючи із себе рідку складову. В ретракції тромбу обов’язкову участь приймають тромбоцити, що виділяють тромбостенін (актоміозиновий комплекс), який активується при контакті з АТФ. Так завершується каскадний процес внутрішнього згортання крові.
Зовнішній механізм згортання крові ініціюється факторами згортання, що виділяються із ушкоджених тканин. При цьому, фактор III (тканинний тромбопластин), взаємодіючи з VII фактором (проконвертином) і іонами Са2+, перетворює фактор VII в активний стан (фактор VIIa), який, у свою чергу, активує фактор Х (фактор Стюарта-Пауера). Активний фактор Х (Ха) в комплексі з фактором V (проакселерином) та іонами Са2+ утворюють фермент, що перетворює протромбін у тромбін.
Далі механізм зовнішнього згортання крові йде по вже відомому шляху внутрішнього згортання аж до утворення стабілізованого фібрину.
Наслідки: утворення тромбу,
розвиток ішемії, інфаркту, гангрени. При відриванні тромбу – розвиток
тромбоемболії.
82. Емболія: визначення поняття, види емболів. Особливості патогенезу емболії великого і малого кіл кровообігу, системи ворітної вени.
Емболія
– переміщення з течією крові і закупорка судин об’єктами (твердими частинками,
рідиною, газами, жиром), яких у нормі не має бути в кровоносних судинах.
Класифікація : а) за характером і походженням : екзогенна (повітряна, газова,
бактеріальна, паразитарна, емболія твердими сторонніми тілами), ендогенна
(тромбоемболія, жирова, тканинна, навколоплідними водами); б) за локалізацією :
емболія великого і малого кола кровообігу, системи ворітної вени; в) рідкі
форми : ретроградна, парадоксальна.
Причини: екзогенна емболія розв в наслідок того, що в судини потрапляє повітря
або гази. Повітряна емб виникає при пораненні великих глибоких вен голови і
шиї. Газова емб при різколу перепаді атмосферного повітря (розгерметизація
літака). Джерелом тромбоемболії є тромб, що відірвався. Жирова емб – при
переломі кісток , що містять всередині білий кістковий мозок. Емболія
навколоплідними водами – потрапляння навколопл вод у пошкодженні судини матки.
1. Емболія великого кола кровообігу: джерелом є тромби, що утв на внутр.
Поверхні порожнин лівої половини серця і в артеріях вел кола кровообігу. Може
виникнути газова і жирова емболія. Закупорка артерій – ішемія – інфаркти.
Розвиток рефлекторного спазму судин веде до розвитку місцевих і загальних
змін(поруш системи гемодинаміки).
2. Емболія малого кола кровообігу: виникає при відриві тромбів, що утв у венах
великого кола кровообігу і на внутр поверхні порожнин правої половини серця.
Емболія судин малого кола веде до порушення зовн дихання і системної
гемодинаміки.
3. Емб системи ворітної вени: джерелом емболів є судини, по яких кров рухається
від внутр. органів до печінки. Результат: розвиток синдрому портальної
гіпертензії.
83. Стаз: визначення поняття, види, причини, патогенез, наслідки.
Стаз – це сповільнення і зупинка течії крові в капілярах, дрібних артеріях і венах.
Розрізняють істинний (капілярний) стаз, який виникає
внаслідок патологічних змін в капілярах або порушення реологічних властивостей
крові, ішемічний – внаслідок повного припинення припливу крові з відповідних артерій
у капілярну сітку, і венозний – як результат венозної гіперемії.
Основними причинами, які
викликають стаз в мікросудинах, є порушення реологічних властивостей крові.
Реологічні властивості крові як неоднорідної рідини мають особливо важливе
значення при її течії по мікросудинах, просвіт яких можна зіставити з величиною
її формених елементів. Внаслідок руху в просвіті капілярів і прилеглих до них
дрібних артерій і вен еритроцити і лейкоцити змінюють свою форму – вигинаються,
витягуються в довжину і т. д. Нормальна течія крові по мікросудинах можлива
тільки, якщо:
· формені елементи можуть легко деформуватися;
· вони не склеюються між собою і не утворюють агрегати, які могли б ускладнювати кровоплин і навіть повністю закупорювати просвіт мікросудин;
· концентрація формених елементів крові не є надлишковою.
Механізм розвитку справжнього стазу пояснюється внутрішньо-капілярною агрегацією еритроцитів, тобто їх склеюванням і утворенням конгломератів, які ускладнюють кровоплин. При цьому підвищується периферичний опір.
Агрегація виникає внаслідок зміни фізичних властивостей плазмолеми еритроцитів під безпосередньою дією факторів, які проникають всередину капілярної судини.
Результати електронно-мікроскопічного вивчення феномена агрегації еритроцитів свідчать про те, що їх поверхня, гладка за нормальних умов, при посиленій агрегації стає нерівною, “пухнастою”, змінюються сорбційні властивості еритроцитів відносно деяких фарбників, що свідчить про порушення їхнього фізико-хімічного стану.
У патогенезі справжнього стазу важливе значення надається сповільненню кровоплину в капілярних судинах у результаті згущення крові.
Провідну роль при цьому відіграє підвищення проникності стінки капілярних судин, розташованих в зоні стазу. Цьому сприяють етіологічні фактори, які викликають стаз, і метаболіти, які утворюються в тканинах.
Особливе значення в механізмі стазу мають біологічно активні речовини (серотонін, брадикінін, гістамін), а також ацидотичне зрушення реакції середовища і його колоїдного стану. Як результат, відзначається підвищення проникності судинної стінки і розширення судин, яке призводить до згущення крові, сповільнення кровоплину, агрегації еритроцитів і, як наслідок, – до стазу. Особливо важливим є вихід у тканини плазмових альбумінів, які сприяють негативному заряду еритроцитів, що може супроводжуватися їхнім випадінням із зваженого стану.
Наслідки стазу. Якщо стаз не довготривалий, то плин крові швидко відновлюється, і в тканинах не встигають розвинутись патологічні процеси. Та при довготривалому стазі розвиваються дистрофічні зміни тканин і некроз.
84. Порушення мікроциркуляції, класифікація. Сладж - синдром: визначення поняття, причини і механізми розвитку. Порушення місцевого лімфообігу, види, причини і механізми розвитку.
Мікроциркуляція, місцевий кровообіг –
кровопостачання окремих органів і тканин. Порушення мікроциркуляції (МЦ)
включає : артеріальна гіперемія, венозна гіперемія, ішемія, стаз,
тромбоз,емболія.
Сладж-синдром – феномен, при якому відбувається прилипання один до одного еритроцитів,
лейкоцитів і тромбоцитів – збільшення в’язкості крові, що утруднює її рух по
мцр.
Причини: є перед станом
стазу. Може настати внаслідок дії отрути, скипидару, фізичних чинників
(температура), токсинів мікроорганізмів.
Механізм: внутрішньо капілярна агрегація еритроцитів/посилення
еритропоезу(поліцитемія)/деформація клітин еритроцитів – згущення крові. Це
все підвищує в’язкість крові, отже збільшує гемодинамічний опір судин.
Порушення лімфообігу – це стан, при якому лімфатичні судини не виконують свою
основну функцію – здійснення постійного й ефективного дренажу інтерстицію.
Розрізняють наступні форми порушення лімфообігу:
1. Механічна недостатність. Проявляється утрудненням відтоку лімфи у зв’язку з наявністю органічних (здавлювання пухлиною, рубцем, облітерація лімфатичних судин при їхньому запаленні, тромбозі та ін.) або функціональних причин (підвищення тиску в магістральних венозних судинах, спазм лімфатичних судин, припинення м’язових скорочень та ін.).
2. Динамічна недостатність. Виникає тоді, коли об’єм транссудації міжтканинної рідини перевищує можливості лімфатичної системи забезпечувати ефективний дренаж інтерстиціальної тканини.
3. Резорбційна недостатність. Зумовлена структурними змінами проміжної тканини, нагромадженням білків і осадженням їхніх патологічних варіантів в інтерстиції.
Основними проявами недостатності лимфообігу в гострій стадії є набряк, нагромадження білків і продуктів їхнього розпаду в інтерстиціальній тканині, а в хронічній стадії – розвиток фіброзу та склерозу.
85. Пухлина: визначення поняття, принципи класифікації пухлин. Загальні закономірності пухлинного росту. Молекулярно – генетичні основи безмежного росту і потенційного безсмертя пухлинних клітин.
Пухли́на — патологічний процес, представлений
новоствореною тканиною, в якій зміни генетичного апарату клітин призводять до
порушення регуляції їхнього росту і диференціювання. Класифікація: доброякісні
і злоякісні пухлини.
Закономірності: 1) пухлинні клітини розмножуються без зовнішнього стимулу до
поділу – мають внутрішні стимули до поділу, не залежні від зовнішніх
регуляторних сигналів; 2) пухлинні клітини здатні ділитись без попереднього
прикріплення до поверхні – ділення «вільного плавання»; 3) у культурі клітин
немає контактного гальмування – розмножуються увесь час, утворюючи багатошарову
структуру; 4) безсмертні – імортилізація – відсутній ліміт Хейфліка(генетично
запрограмована кількість поділів,яку може здійснити кл), тобто кількість
поділів – безмежна.
86.Типові властивості доброякісних та злоякісних пухлин. Види анаплазії. Шляхи й механізми метастазування.
Властивості:
1.Доброякісні пухлини складаються з добре диференційованих клітин. Ці пухлини зберігають типову структуру тканини, з якої утворилися. У той же час злоякісні пухлини характеризуються втратою диференціювання клітин, спрощенням і ати-повістю своєї будови.
2. Доброякісні пухлини часто ростуть повільно, їх ріст може зупинитися, а іноді спостерігається й зворотний розвиток (регресія). Для злоякісних пухлин, як правило, характерний швидкий ріст, що спонтанно не зупиняється. Мимовільна регресія таких пухлин невідома.
3. Доброякісні пухлини мають капсулу й ростуть експансивно, тобто не проростають у навколишні здорові тканини, а розсовують їх. Ріст злоякісних пухлин інвазивний (інфільтративний). Вони не мають капсули і проростають у навколишні тканини.
4. Доброякісні пухлини не метастазують, у той час як злоякісні зазвичай дають метастази.
5. Доброякісні пухлини добре лікуються хірургічними методами, летальних наслідків, як правило, не буває. Злоякісні пухлини при відсутності лікування призводять до смерті.
Анаплазія - набуття пухлинними клітинами властивостей, характерних для ембріонального етапу розвитку організму. Інакше кажучи, пухлинні клітини своїми структурними, функціональними, біохімічними, імунологічними властивостями нагадують ембріональні клітини.
ВИДИ АНАПЛАЗІЙ:
1.Морфологічна -зводиться до появи тканинної, клітинної і
субклітинної атиповості .
2.Біохімічна-це особливості метаболізму пухлинних клітин ,
викликані змінами їх генетичного апарату.
Найхарактерніші біохімічні особливості пухлинних клітин
стосуються обміну білків і вуглеводів. Синтез білків переважає
над їх розпадом. Для побудови власних білків пухлина
захоплює амінокислоти з інших органів, в пухлині активується
анаеробний гліколіз і ферменти , які його забезпечують.
3.Фізико-хімічна - серед фізико-хімічних особливостей пухлинних
клітин слід виділити такі :
а) ацидоз - виникає внаслідок нагромадження молочної кислоти;
б)внутрішньоклітинну гідратацію;
в) нагромадження іонів К ;
г)підвищення електропровідності ;
д)зменшення їх поверхневого натягу;
4.Імунна - це зміни антигенних властивостей пухлинної клітини. Ці зміни
– результат перебудови білкового обміну. Відомо , що кожна
тканина синтезує специфічний для неї набір антигенів. У пухлині
цей набір змінюється.
5.Функціональна-- проявляється втратою або спотворенням виконуваної
клітинної функції.
Шляхи і механізми метастазування.
Метастазування — це здатність злоякісної пухлини давати вторинні пухлинні вузли (метастази) у віддалених від первинної пухлини частинах організму.
Механізм метастазування складається з трьох етапів.
І. Вихід пухлинних клітин із тканини в лімфатичні або кровоносні судини - інтравазація. Цьому передує прикріплення пухлинних клітин до компонентів базальної мембрани судин. В основі такого прикріплення лежить утворення "ламінінових містків" між поверхнею клітин пухлини і колагеном IV типу базальної мембрани. Глікопротеїд ламінін є компонентом інтерстиціальної речовини базальної мембрани. Деякі найбільш злоякісні клітини самі здатні продукувати цю речовину. Ламінін, з одного боку, взаємодіє з так званими ламініновими рецепторами пухлинних клітин, а з другого - з хімічними групами колагену IV типу.
II. Транспорт пухлинних клітин лімфою й кров'ю - дисемінація. Із клітин пухлини, що проникли в судини, утворюються пухлинні емболи. їх утворення пов'язане з агрегацією пухлинних клітин. У цьому процесі обов'язково беруть участь тромбоцити.
III. Вихід пухлинних клітин з судин у тканини й утворення вторинних вогнищ (метастазів)— екстравазація. Для локалізації метастазів мають значення місцезнаходження первинної пухлини, шлях метастазування (лімфогенний чи гематогенний), рецепторна взаємодія пухлинних клітин з ендотелієм судин.
Три шляхи метастазування пухлинних клітин:
1— гематогений — по кровоносних судинах;
2— лімфогений — по лімфатичних судинах;
3— тканинний — безпосередньо від однієї прилеглої тканини до іншої або по міжтканинних просторах.
Частіше за все метастазування відбувається лімфогенним шляхом. Проте тканина, в яку потрапив метастаз, повинна бути здатною сприйняти цей метастаз.
87. Етіологія пухлин. Загальна характеристика канцерогенів. Фактори ризику (генетичні/хромосомні дефекти, аномалії конституції) і умови виникнення та розвитку пухлин.
Етіологічні фактори, що викликають розвиток злоякісних пухлин називають канцерогенами. Виділяють три групи канцерогенів:
· фізичні (іонізуюча радіація, ультрафіолетове випромінювання, висока температура, механічний вплив);
· біологічні (онкогенні віруси);
· хімічні (канцерогенні речовини) : речовини, здатні викликати розвиток злоякісних пухлин.
За походженням розрізняють канцерогени природні і штучні. її. За хімічною
будовою канцерогени можуть бути представлені:
а) поліциклічними ароматичними вуглеводнями (ПАВ);
б) ароматичними амінами;
в) нітрозосполуками;
г) мікотоксинами;
ґ) гетероциклічними вуглеводнями;
д) аміноазосполуками;
є) простими сполуками (миш'як, азбест та ін.).
III. Відносно організму хімічні канцерогени можуть бути екзогенними й ендогенними.
IV. За механізмом канцерогенного впливу розрізняють канцерогени прямої дії й канцерогени непрямої дії (проканцерогени).
До канцерогенів природного походження відносять продукти життєдіяльності деяких грибів (мікотоксини) і продукти вулканічної діяльності.
Джерелами канцерогенів штучного походження є:
1) викиди промислових підприємств;
2) вихлопні гази автомобілів;
3) тютюновий дим;
4) продукти неправильної кулінарної обробки їжі (використання пересмажених жирів, порушення технології копчення та ін.).
Фактори ризику:
І. Ендогенні фактори
1. Пов’язані з репродуктивною функцією. а. менструальна, сексуальна, репродуктивна та лактаційна;
2. Фактори пов'язані з супутніми та майбутніми захворюваннями;
3. Генетичні та спадкові фактори.
ІІ. Екзогенні фактори
1. Пов'язані зі способом життя та професією (харчування, гіподинамія).
Розвиток пухлини визначається не тільки властивостями самих пухлинних клітин, але й впливом організму на цей процес. Найбільше значення мають:
а) васкуляризація пухлини. Показано, що максимальна віддаленість пухлинних клітин від просвіту кровоносних судин не може бути більше 1—2 мм. Якщо відстань перевищує цю величину, клітини пухлини гинуть. У злоякісну пухлину, як правило, інтенсивно проростають кровоносні судини. Це пов'язане з тим, що пухлинні клітини вивільнюють так званий ангіогенетичний фактор (ангіогенін), що стимулює ріст капілярів і розмноження ендотеліальних клітин;
б) гормони. Хоча пухлинний процес є автономним, існують, однак, пухлини, які виявляють високу чутливість до гормонів. Це рак молочної залози, матки, яєчників, передміхурової залози. Одні гормони посилюють ріст зазначених пухлин, інші гальмують;
в) стан механізмів протипухлинного захисту організму.
88. Фізичні канцерогени. Хімічні канцерогени: принципи класифікації, характеристика основних груп. Механізми хімічного канцерогенезу. Коканцерогенез та синканцерогенез. Органотропність канцерогенів.
Серед факторів фізичної природи до етіології пухлин можуть мати стосунок:
1) іонізуюча радіація;
2) ультрафіолетове випромінювання;
3) механічний вплив (тривалий тиск на тканини);
4) висока температура.
Роль іонізуючоїрадіації доводять в експериментах і численними клінічними спостереженнями. Радіаційний канцерогенез було виявлено вже через 7 років після відкриття рентгенівського випромінювання. Першою його жертвою став перший виробник рентгенівських трубок Фрікен, що перевіряв якість своєї продукції на власних руках. У нього виник рак шкіри.
При тривалому перебуванні білих щурів на сонці у них часто розвивається рак шкіри, що пов'язують із впливом ультрафіолетового випромінювання.
Розвиток пухлини в ділянці імплантації тваринам пластинок, виготовлених з хі-Вачно інертного матеріалу ("пластмасовий " канцерогенез), виникнення раку в місці тривалого тиску на шкіру кантрі (спеціальної сумки, у якій жителі Індії носять вугілля) - це приклади можливого значення механічних факторів у канцерогенезі.
Нарешті, часте виникнення пухлин порожнини рота у пастухів гірських районів, які п'ють дуже гарячий чай, наводить на думку про можливу роль високої температури в розвитку пухлин.
Хімічні канцерогени — це речовини, здатні викликати розвиток злоякісних пухлин. ї. За походженням розрізняють канцерогени природні і штучні. За хімічною будовою канцерогени можуть бути представлені:
а) поліциклічними ароматичними вуглеводнями (ПАВ);
б) ароматичними амінами;
в) нітрозосполуками;
г) мікотоксинами;
ґ) гетероциклічними вуглеводнями;
д) аміноазосполуками;
є) простими сполуками (миш'як, азбест та ін.).
III. Відносно організму хімічні канцерогени можуть бути екзогенними й ендогенними.
IV. За механізмом канцерогенного впливу розрізняють канцерогени прямої дії й канцерогени непрямої дії.
Характеристика канцерогенної дії хімічних сполук
Поліциклічні ароматичні вуглеводні. Поліциклічні ароматичні вуглеводні - це хімічні сполуки, що виявляються у нафтопродуктах та інших природних багатокомпонентних сумішах. Значна частина ПАВ є хімічними канцерогенами, що індукують злоякісні пухлини молочних залоз, м‘язової та сполучної тканини. До типових ПАВ відносяться 7,12-диметилбензантрацен та бенз (а) пірен (БП), 20-метилхоланрен, 1,2, 5,6-дибензантрацен, а також сполуки, що мають гетероциклічні атоми азоту, наприклад 9-метил-3,4-бензакридин та 4-нітрохінолін-N-оксид.
ПАВ добре вивчені на прикладі БП- індикаторної сполуки цієї групи канцерогенів. БП характеризується максимальною відносною стабільністю при різноманітних фізико-хімічних впливах. Він завжди визначається там, де присутні і інші канцерогенні вуглеводні, будучи одним з найбільш розповсюджених та сильних канцерогенних агентів.
До природних абіогенних джерел, що формують природній фон ПАВ, відносять вулканічну діяльність, процеси нафто-, вугле- та сланцеутворення. Встановлена можливість синтезу ПАВ рослинними організмами (зокрема, декотрими злаковими), рядом бактерій (наприклад, Clostridium putride), фітопланктоном.
У результаті діяльності людини забрудненість біосфери канцерогенними ПАВ набагато збільшилась, а у промислових районах в сотні та тисячі разів перевищує їх природний фоновий рівень. Основні антропогенні джерела забруднення ПАВ атмосфери- промислові викиди та вихлопні гази автомашин.
Наявність ПАВ у викидах турбореактивних двигунів літаків є причиною широкого розповсюдження цих речовин у всіх шарах біосфери. Циркуляція ПАВ в атмосфері залежить від дисперсних часточок, котрими вони сорбуються, ступеню віддаленості джерела ПАВ від поверхні Землі, інтенсивності сонячної радіації, наявності природних фотооксидантів, що сприяють руйнуванню БП та інших канцерогенних ПАВ. Деструкція канцерогенних ПАВ може відбуватися під впливом УФ променів та озону.
Забруднення ПАВ водних екосистем відбувається у результаті скиду промислових стічних вод, а також викидів двигунів річних та морських кораблів. Циркуляція ПАВ у водному середовищі включає у себе процес їх розподілення між різними компонентами водної екосистеми та включення у ланцюги харчування. Ці процеси сприяють деструкції та зниженню вмісту ПАВ у водоймі. Більша частина ПАВ, як і більшість хімічних речовин, сорбується зваженими частками та осідає з ними на дно, звідки надходить у водорості та вищі водні рослини (водорості кумулюють більше ПАВ, ніж вищі водні рослини). Менша частина ПАВ, розчинена у воді, накопичується у мікропланктоні, по мірі відмирання котрого надходить у донні відклади.
Як у прісних, так і у морських водоймах виявлена мікрофлора, що здатна трансформувати БП. Специфічна біодеградація ПАВ за участю ендокринних систем клітин іде як у рослинних, так і у тваринних організмах (зокрема, у рибах). В організмі молюсків відсутні ендокринні системи деструкції ПАВ, внаслідок чого молюски накопичують канцерогенні речовини.
БП, що накопичуються у верхніх шарах (до 3см) ґрунту, підлягають деструкції, швидкість котрої залежить від кількості БП, pH та вологості ґрунту, але у першу чергу- від складу мікробіоценозу. Біологічна очистка ґрунту від ПАВ забезпечується декотрими бактеріями, широко розповсюдженими у забрудненому ПАВ ґрунті та воді. Частково ПАВ накопичуються у рослинах.
Ароматичні азосполуки. Більшість ароматичних азосполуки відносяться до азофарбників. Для них є характерною наявність двох або більше азогруп. В залежності від цього вони можуть поділятися на моно-, ди-, три- та більше азопохідні. Азофарбники застосовують для фарбування натуральних та синтетичних тканин, для кольорового друку у поліграфії, у косметиці, кольоровій фотографії і т. ін. До них відносяться моноазобензол, N, N’-диметил-4-аміноазобензол.
Канцерогенна дія азосполук виявляється як при введенні їх тваринам у травневий тракт, так і при підшкірному введенні та навіть змащуванні шкіри. Пухлини при цьому виникають не на місці введення канцерогенів, а в органах, віддалених від місця аплікації (печінка, сечовий міхур). Встановлено різний ступінь канцерогенної активності речовин у залежності від їх хімічної структури. Водорозчинні та сірковмісні азосполуки, як правило, не є канцерогенними.
Ароматичні аміносполуки. До цієї групи сполук відносяться речовини, що мають структуру або дифенілу або нафталіну (4, 4’-диамінодифеніл, 4, 4’-диаміностильбен, 2-нафтиламін), також флуорен, що є похідним дифенілу.
Ароматичні аміносполуки знаходять широке застосування у різноманітних областях промисловості (вони використовуються як напівпродукти при синтезі органічних сполук, фарбників та особливо бензиденових фарбників, також можуть застосовуватись для синтезу лікарських препаратів, інсектицидів і т. ін.). Для канцерогенних сполук характерною є наявність однієї або двох аміно-, а можливо, і нітрогруп, що розміщуються у пара положенні та приєднаних до ароматичних систем незалежно від характеру зв‘язку між бензольними кільцями при умові досить міцного з‘єднання [Плісс Г. Б., 1965].
Нітрозосполуки. Нітрозосполуки - це хімічні сполуки, що мають одну або декілька нітрогруп (NO-), зв‘язаних з атомами вуглецю. Вони відносяться до найбільш небезпечних канцерогенів, що викликають злоякісні пухлини печінки, легень, нирок, травневого тракту у 40 видів тварин різних класів
До найбільш вивчених нітрозосполук, що проявляють канцерогенну активність, відносяться N-нітрозодиметиламін, N-нітрозодиетиламін, N-метил-N-нітрозогуанідин, нітрозодиметилсечовина. Декотрі нітрозосполуки застосовуються у клініці як протипухлинні засоби. Нітрозосполуки утворюються при виробництві гуми, азофарбників та риб‘ячої муки, а також при смаженні їжі і особливо при курінні тютюну.
Ряд нітрозосполук є продуктами метаболіту вищих та нищих рослин. Вони можуть утворюватися у повітрі на поверхні часточок аерозолю, а також на поверхні епітелію дихальних шляхів з оксидів азоту та летючих амінів. В утворенні нітрозосполук приймають участь і мікроорганізми, що забезпечують кругообіг азоту у атмосфері, а також представники ендогенних мікробів- декотрі штами Escherichia coli та ентерококів.
Попередниками нітрозосполук є формальдегід та інші альдегіди, галогеноїди, кетони, поверхнево-активні речовини. Важливе значення в утворенні нітрозосполук має мікрофлора води (в гідроценозах), а також мікрофлора кишківника людини (ендогенний синтез нітрозосполук). Оскільки в утворенні та циркуляції нітрозосполук приймають участь мікроорганізми, вищі та нищі рослини, їх відносять до біогенних канцерогенів.
Основним джерелом забруднення ґрунту є промислові та побутові стічні води та добрива. Серед останніх найбільш небезпечними є різноманітні комбінації мінеральних добрив та пестицидів, а також забруднення типу формальдегідів та фенольних сполук. Надлишкове незбалансоване застосування мінеральних добрив (зокрема азотних) призводить до зросту забруднення їх рослинних продуктів як нітрозосполуками, так і їх попередниками. Відмічено збільшення вмісту нітратів у круп‘яних та зернових продуктах по мірі збільшення строку та у залежності від умов зберігання. Забрудненість харчових продуктів тваринного походження, як правило, обумовлене умовами їх технологічної обробки.
Характерною особливістю нітрозосполук є можливість їх ендогенного синтезу з вторинних та третинних амінів та нітратів. Ендогенний синтез нітрозосполук відбувається з попередників (нітритів, нітратів, амінів та амідів), що надходять у їжу людини та тварин у вигляді консервантів або у складі рослинних продуктів. Нітрати можуть накопичуватись у рослинах у результаті застосування надлишкової кількості добрив, що забруднюють ґрунт та водойми. Джерелом амінів є білкові продукти харчування, вони є у овочах, чаї, хлібі, у декотрих лікувальних препаратах. Виявлені нітрозосполуки у багатьох продуктах хімічної та фармацевтичної промисловості. Висока забрудненість нітрозосполуками косметичних засобів, а також порошків та розчинів для миття посуду та чистки поверхонь.
Таким чином, розповсюдження нітрозосполук та їх попередників відбувається в основному через рослинні продукти харчування, питну воду, їжу тварин та продукти тваринного походження. Так, 10-20% нітратів надходить в організм людини з м‘ясними продуктами, а решта головним чином з овочами. Нітрозація лікувальних засобів, що є вторинними або третинними амінами, також легко відбувається ендогенним шляхом. Серед 60 досліджених лікувальних засобів нітрозації підлягають амідопірин, анальгін, бромгексин, ампіцилін, іміпрамін, оксацилін, клоліпрамін, дезипрамін, етамбутол, окситетрациклін, піперазин.
Для обмеження надходження канцерогенних нітрозосполук у організм людини та тварин рекомендується обмежити їх надходження ззовні шляхом нормування вмісту нітратів та нітритів у їжі, кормах та воді. Ефективним профілактичним засобом є аскорбінова кислота, що інгібує утворення нітрозосполук у організмі, та галова кислота.
Метали, металоїди, азбест. В експериментах на тваринах вдалось довести, що ряд металів та металоїдів мають канцерогенну активність. До них відносяться нікель, хром, миш‘як, кадмій, берилій, кобальт, свинець, титан, цинк, залізо. В наш час є епідеміологічні дані, що свідчать про канцерогенну небезпеку для людини ряду технологічних процесів та робіт, пов‘язаних з очисткою та збагаченням нікелю, видобуванням гематиту (диоксиду заліза), контактом з хромом (ÚI) та його солями, миш‘яком.
За сучасними уявленнями, свою канцерогенну дію на клітини тканин метали здійснюють або шляхом безпосереднього впливу на генетичний апарат клітини, або через ферментативні системи, каталізуючи біологічне окислення, синтез нуклеїнових кислот та білків. Гіпотеза прямої дії на спадковий апарат клітини допускає, що метали, знаходячись в організмі у надлишку, осідають на поверхні хромосом, індукуючи при цьому хімічні та електрохімічні процеси, що викликають зміни в структурі нуклеїнових кислот [В. Вакео, Е. Халме, 1969].
Що ж стосується конкретної класифікації і характеристики генотоксичної та канцерогенної дії металів, то в цьому напрямку Міжнародна Агенція Вивчення Раку (МАВР) провела систематизацію накопичення знань із урахуванням клінічних, епідеміологічних та експериментальних досліджень. Усі канцерогенні метали для людини і тварин розділені на групи:
Перша група- безумовно канцерогенні для людини: хром.
Друга група А- можливо канцерогенні для людини: нікель, берилій.
Друга група В- можливо канцерогенні для людини: кадмій, цисплатина.
Третя група- канцерогенні для тварин: кобальт, марганець, цинк, титан, свинець, кадмій, залізо, магній, сурьма, олово, хром, миш’як.
Таким чином, нині твердо встановлена канцерогенність п’яти металів для людини та 10-12 металів для тварин.
Дані експериментальних та клінічних спостережень засвідчують, що окремі метали, маючи канцерогенну дію, у певних дозах можуть гальмувати експериментальні та клінічні форми канцероматозу.
Інгібування сформованого злоякісного росту металами за механізмом дії реалізується двома принципово різними шляхами: гальмівним впливом на злоякісний поділ клітин через підсилення опірності протипухлинної дії гомеостазу організму у цілому та специфічною канцероцидною дією їх на ракові клітини шляхом блокування енергозабезпечення, порушення структури та функції біологічних мембран. Більшість металів, що проявляють інгібуючий вплив на злоякісний ріст (кобальт, марганець, цинк, мідь, селен, миш’як), реалізують цю дію через вплив на активність ферментів, гормонально-вітамінний, мікроелементний баланс, мобілізуючи опірність гомеостазу, пухлинному процесу [1].
Значне місце у виникненні професійного раку людини займає азбест. Азбест відноситься до природних силікатних матеріалів силікатної структури з характерним рядом цінних властивостей (стійкість до високих температур, до хімічних агентів, здатність підлягати обробці на ткацьких верстатах і т. ін.). В залежності від фізико-хімічних властивостей він поділяється на хризотил (білий азбест) та амфібол. Останній у свою чергу, поділяється на крокідоліт (голубій), тремоліт, антофіліт, амозит, магензиаарфведсоніт, актиноліт та інші.
Встановлено, що при подальшому контакті у робочих, що займаються видобуванням та переробкою азбесту, виникають пухлини легень та мезотеліоми плеври. Описані також виникнення пухлин шлунково-кишкового тракту та розвиток мезотеліоми очеревини [Selikoff J., Lee D., 1978]. Визначено, що бластомогенна активність азбесту залежить від розмірів волокон: найбільш активні волокна з довжиною не менше 7-10мкм та товщиною не більше 2-3мкм.
У хімічному канцерогенезі виділяють три стадії: ініціацію, промоцію і пухлинну прогресію.
Сутність ініціації полягає в тому, що під впливом хімічного канцерогену відбувається
трансформація клітини, тобто перетворення її з нормальної на пухлинну.
Під час стадії промоціїтрансформована клітина отримує стимул до розмноження, пухлина починає рости. Речовини, що стимулюють розмноження трансформованих клітин, отримали назву промоторів. Самі вони можуть бути й неканцерогенни-ми. Дуже сильними промоторами є форболові ефіри, які активують протеїнкіназу С, що бере участь у регуляції клітинного поділу.
Між ініціацією і промоцією може пройти багато часу (іноді роки). Більшість вивчених хімічних канцерогенів є повними, тобто виявляють властивості й ініціаторів, і промоторів.
Пухлинна прогресія — це якісні зміни властивостей пухлини в процесі її розвитку. Як правило, згодом пухлина набуває усе більш і більш злоякісних властивостей.
Сінканцерогенез - спільна дія декількох канцерогенів і суммация їх ефектів. Коканцерогенез - посилення дії канцерогенів факторами, які не є самі канцерогенами
.
89. Біологічні канцерогенні фактори. Класифікація онкогенних вірусів. Вірус-індуквані та вірус-асоційовані пухлини у тварин і людей. Механізми вірусного канцерогенезу.
До онкогенних ДНК вмісних вірусів відносять:
а) nanoea-eipycu. Вони викликають розвиток у тварин трьох видів пухлин: папіломи, поліоми і пухлини, що виникає під дією вакуолізуючого вірусу мавп SV-40;
б) аденовіруси. Онкогенними для тварин є аденовіруси 12,18 і 31-го типів;
в) герпес-віруси, зокрема, вірус Епшгпейна — Барр.
Що стосується людини, то в одних випадках доведено, а в інших є підстави думати, що причиною деяких пухлин є:
а) вірус Епштейна - Барр (викликає лімфому Беркіта, назофарингеальний рак);
б) вірус гепатиту В (може бути причиною раку печінки);
в) вірус папіломи людини (викликає доброякісні пухлини шкіри, жіночих геніталій, гортані).
РНК-вмісні віруси є онкогенними для тварин і людини
Всі онкогенні РНК-вмісні віруси належать до сімейства pempoeipycie. Загальною їхньою властивістю є наявність у вірусному геномі гена, що кодує структуру ферменту ревертази, відомого ще як зворотна транскриптаза, або РНК-залежна ДНК-полімераза. Цей фермент забезпечує синтез двоспіральної ДНК на матриці односпіральноїРНК. Унаслідок цього утворюється ДНК-копія ретровірусу, що отримала назву ДНК-провірусу.
Залежно від онкогенності ретровіруси поділяють на дві групи.
I. Гостротрансформуючі ретровіруси. Дуже онкогенні, викликають розвиток пухлин після короткого латентного періоду. Ці віруси мають у своєму геномі онкоген, тому в основі трансформації клітин у пухлинні лежить епігеномнш механізм. До зазначеної групи, зокрема, відносять віруси гострих лейкозів у птахів, мишей і саркоми Рауса у курей.
II. Повільнотрансформуючі ретровіруси. Викликають розвиток пухлин після тривалого латентного періоду. Ці віруси не мають у своєму складі онкогена, тому основний механізм їх трансформуючої дії — мутаційний. До вірусів цієї групи відносять віруси лімфолейкозів.
Онкогенним ретровірусом людини є вірус Т-клітинноїлімфоми-лейкемії. Він передається від людини до людини при тривалих інтимних контактах, переливанні крові. Слід зазначити, що цей лімфотропний вірус дуже схожий на віруси імунодефіциту людини (ВІЛ), що викликають СНІД.
етапи вірусного онкогенезу.
I. Рецепція вірусу. Відбувається взаємодія вірусної частки з певними структурами плазматичної мембрани клітини (рецепторами). Відсутністю відповідних рецепторів пояснюється видовий імунітет до вірусної інфекції.
II. Роздягання й проникнення вірусу в клітину (інтернолізація).
III. Об'єднання (інтеграція) вірусного геному з геномом клітини. Це центральний і обов'язковий етап вірусного онкогенезу. У випадку ДНК-вмісних онковірусів відбувається вбудовування вірусної ДНК у ДНК клітини; у випадку РНК-вмісних вірусів - вбудовується ДНК-провірус, який утворюється під впливом ферменту ревертази.
IV. Постійне перебування (персистенція) вірусу в геномі клітини. При цьому вірус розмножується разом з клітиною. Такий перебіг вірусної інфекції називається абортивним. Абортивний перебіг є неодмінною умовою перетворення клітини в пухлинну під впливом вірусу.
V. Трансформація клітини.
VI. Промоція.
VII. Пухлинна прогресія (див. запит. 16.19 і 16.20).
фактори, від яких залежить трансформуюча дія вірусів на клітину.
I. Фактори, що їх визначають властивості вірусу. До них, зокрема, відносять так звану структурну дефектність вірусу. Структурно дефектними називаються віруси, які в процесі розмноження й формування вірусних часток втратили частину свого геному. Вони здатні проникати в клітину й об'єднуватися з її геномом, однак не здатні до репродукції. У цьому випадку єдино можливим варіантом вірусної інфекції є абортивний її перебіг. Це збільшує ймовірність трансформації клітини під впливом структурно дефектних вірусів.
Крім того, для механізмів онкогенної дії вірусів має значення наявність або відсутність у їхньому геномі вірусного онкогена.
II. Фактори, що їх визначають властивості клітин. До них відносять наявність віщіовіщтх. рецепторів на поверхні клітин, період клітинного циклу, під час якого вірус потрапляє в клітину. Інтеграція геному вірусу з геномом клітини можлива тільки в синтетичну фазу (фазу S) або в період, що безпосередньо передує їй (кінець фази G,). Цим, зокрема, пояснюється та обставина, що високодиференційова-ні клітини, які втратили здатність до поділу, є стійкими до дії онкогенних вірусів. Крім того, клітина стає чутливою до онкогенних вірусів, якщо в ній нема умов для синтезу всіх білків, необхідних для формування вірусних часток. За таких умов неможлива репродукція вірусу й вірусна інфекція має абортивний перебіг, при якому збільшується ймовірність трансформації клітини. Таке явище отримало назву функціональної дефектності вірусу.
молекулярні механізми в основі вірусного онкогенезу
I. Продукти вірусного онкогена порушують регуляцію клітинного поділу й у такий спосіб викликають трансформацію клітини. Цей механізм отримав назву епігеном-ного. Він реалізує себе у випадку дії вірусів, що містять у своєму геномі онкогени. Із цим механізмом, зокрема, пов'язаний вплив на клітини гостротрансформуючих ретровірусів.
II. Під дією промоторів і підсилювачів вірусів, що потрапили в геном клітини, відбувається активація власних протоонкогенів, і вони перетворюються на клітинні онкогени, що трансформують клітину. Це так звання мутаційний механізм вірусного канцерогенезу. Він виявляє себе при дії вірусів, позбавлених онкогенів, зокрема, повільнотрансформуючих ретровірусів
90. Патогенез пухлинного росту. Роль порушень молекулярних (генетичних) механізмів регуляції клітинного поділу в процесі пухлинної трансформації. Способи перетворення протоонкогенів на онкогени. Властивості онкобілків.
Вірусні онкогєни - це гени вірусу, з функціонуванням яких пов'язане перетворення нормальних клітин у пухлинні. Білки-продукти вірусних онкогенів порушують регуляцію процесів клітинного поділу і в такий спосіб викликають трансформацію клітини (епігеномний механізм канцерогенезу).
Протоонкогєни - це власні гени клітин, які несуть інформацію про структуру білків, що беруть участь у регуляції клітинного поділу. Протоонкогени є клітинними аналогами вірусних онкогенів.
Вважають, що вірусні онкогєни являють собою протоонкогени, які потрапили в геном вірусів у результаті тривалої еволюції останніх. Це пояснюється тим, що віруси, проходячи через клітини, можуть "красти", тобто прихоплювати із собою клітинні гени.
Протоонкогени є у всіх клітинах. Однак в одних клітинах вони все життя репресовані, тобто "мовчать", в інших вони "працюють" тільки в період ембріогенезу, а в інших - функціонують відповідно до регуляторних сигналів, що надходять.
Як класифікують вірусні онкогени і протоонкогени?
Залежно від продуктів, інформацію про які несуть онкогени вірусів і протоонкогени клітин, їх поділяють на такі групи (рис. 56):
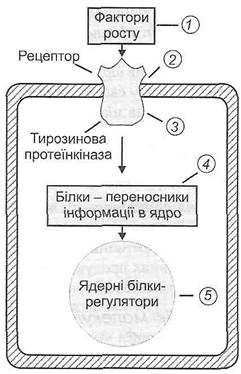
Рис. 56. Продукти протоонкогенів і вірусних онкогенів
1. Онкогени, що кодують фактори росту або їхні аналоги. Наприклад, sis-онкоген вірусу саркоми мавп і аналогічний протоонкоген клітин кодують структуру фактора росту тромбоцитар-ного походження.
2. Онкогени, що кодують клітинні рецептори до факторіє росту.
Наприклад, онкоген вірусу лейкозу курей erb-B несе інформацію про видозмінений рецептор до епідермального фактору росту.
3. Онкогени, що кодують тирозинспецифічні про-теїнкінази. Прикладом може бути scr-онкоген вірусу саркоми Рауса та відповідний протоонкоген клітин.
4. Онкогени, що кодують структуру білків, які переносять інформацію від рецепторів плазматичної мембрани клітини до ядра. Прикладом є ras-онкоген вірусу саркоми щурів, який несе інформацію про цитоплазматичний білок, що зв'язує ГТФ.
5. Онкогени, що кодують структуру ядерних білків-регуляторів.
До них, зокрема, належить mic-онкоген вірусу мієлоцитозу курей. Продукти вірусних онкогенів є аналогами продуктів протоонкогенів, тобто власних білків клітин, що беруть участь у регуляції клітинного поділу. Однак є деякі, хоча й невеликі, відмінності, Вони стосуються певних структурних характеристик вірусних онкогенів і протоонкогенів. Саме ці відмінності і роблять продукти вірусних онкогенів власне онкогенними для клітини.
Що таке клітинні онкогени? Назвіть основні механізми перетворення протоонкогенів у клітинні онкогени.
Клітинні онкогени (трансформуючі гени) - це протоонкогени, які набули здатності трансформувати клітину, тобто перетворювати її в пухлинну. Перенесення таких генів в інші, здорові клітини викликає трансформацію останніх.
Нині доведено існування таких механізмів перетворення протоонкогенів у клітинні онкогени.
І. Дерепресія протоонкогена. Може відбуватися або в результаті порушення структури, а отже, і функції відповідного антионкогена, або внаслідок мутацій у генах-репресорах, що блокують діяльність (експресію) протоонкогена.
її. Підвищення експресії протоонкогена. Спостерігається тоді, коли білок-про-дукт протоонкогена утворюється і в нормі, однак його дуже мало. Під впливом певних генетичних факторів утворення такого продукту іноді істотно зростає. Зазначене явище може бути обумовлене такими конкретними механізмами:
а) ампліфікація генів (збільшення кількості їхніх копій);
б) хромосомні мутації — транслокація;
в) вплив вірусних промоторів і підсилювачів (так діють ретровіруси, що не мають у своєму складі онкогена);
г) вплив мігруючих генів самої клітини (транспозонів).
Ш. Якісні зміни протоонкогена, що викликають утворення видозміненого продукту. Ці зміни, як правило, обумовлені точковими мутаціями протоонкогена.
16.33. Що таке антионкогени?
антионкогени — це клітинні гени, продукти яких викликають репресію прото-онкогенів.
Втрата антионкогенів (делеція) або мутації в них, що призводять до утворення неактивних продуктів, можуть мати своїм наслідком дерепресію протоонкогенів і трансформацію клітини, тобто утворення злоякісної пухлини.
91.Пухлинна прогресія:визначення поняття,причини і механізми,типові ознаки.Механізми інвазійного росту і метастазування.Набуття резистентності до хіміопрепаратів.Вплив пухлини на організм.Патогенез та прояви ракової кахексії.
Прогресія — третій етап механізму канцерогенезу. Стійкі якісні зміни властивостей пухлини, переважно у бік малігнізациї, що виникають під дією декількох чинників: 1. У первинний канцерогенез, залучається декілька клітин, що сприяє формуванню в пухлині, що розвивається, декількох субліній. У зростаючій пухлині постійно здійснюється відбір найбільш життєздатних клітин. Певні клітини отримують перевагу. При зростанні пухлинної тканини в організмі змінюється гормональна регуляція, можливе вироблення антитіл проти клітин, наявних вякій-небудь сублінії. В результаті отримує перевагу яка-небудь з субліній пухлинних клітин, яка спочатку складала меншість. 2. Зміна генотипу і фенотипу клітин, що приводить до прогресії, може бути пов'язане з продовженням дії на геном пухлинних клітин канцерогенного чинника. 3. Спонтанні мутації пухлинних клітин при зниженні в них активності репаратівних ферментів. 4. Надбання пухлинними клітинами нових властивостей, пов'язаних з суперінфекцією пухлиногенними і непухлиногенними вірусами, полегшеною в пухлинних клітинах.
Ріст пухлини може бути експансивним і
інфільтративним. При експансивному рості оточуюча здорова тканина у міру
зростання пухлини розсовується, при інфільтративному — пухлинні клітини
проростають між нормальними клітинами і через судинну стінку. Потрапляючи в
лімфу або кров, вони переносяться в інші органи і можуть утворювати нові
вогнища пухлинного росту (метастази). Експансивний ріст характерний для
доброякісних пухлин, а інфільтрірующий з утворенням метастазів — для злоякісних
пухлин.
Метастазування складається з наступних етапів:
1. Початковий етап — припинення утворення міжклітинних контактів, зміна
рецепторів мембрани і придбання рухливості із-за зміни білків цитоскелета, їх
фосфорилування протпеїнкиназами. Відбуваються зміни регуляції генів, що кодують
білки цитоскелета і рецептори мембран.
2. У клітинах, що трансформуються, відбувається синтез активатора плазміногена.
У пухлинних клітинах утворюються коллагенази. Пухлинні клітини, що не володіють
плазміногеном, виробляють чинник, що залучає моноцити, ферменти яких
розріджують матрикс і створюють можливість пухлинним клітинам метастазувати.
3. Катепсини є як вбудовані в мембрани пухлинних клітин, так і у вільному стані
в міжклітинній рідині пухлинної тканини.
4. Пухлинні клітини володіють набором чинників, що активують функції
сполучно-тканинних клітин по синтезу колагену, глікопротеїнів і інших
компонентів основної речовини.
5. Пухлинні клітини виділяють ангіогенін і інші чинники росту судин, що
забезпечує кровопостачання пухлинної тканини.
6. У мембранах пухлинних клітин радикали нейрамінової кислоти, глікопротеїдів
α-Д-глюкопиранозида і N-ацетил-Д-галактозамінузалишаються відкритими.
Білок конканавалін А, а також лектіни, завдяки наявності відкритих радикалів,
аглютинують пухлинні клітки.
Основні
ланки патогенезу ракової кахексії.
- Збільшені енерговитрати в спокої, за рахунок збільшеного термогенезу в
скелетних м’язах внаслідок експресії розщеплених (uncoupling) протеїнів та
посиленого циклу Ropi.
- втрати жирової тканини – збільшений ліполізис продуктами пухлини і
господаря.
- втрати скелетних м’язів депресія синтезу протеїнів деградація
протеїнів.
- деградація протеїнів збільшена активність убіквітин – протеасом та
лізосом.
- зменшений синтез білків – зменшений рівень фактору 4F, зв’язування
метіонил+РНК до 40S рибосомальної підодиниці, збільшення фосфорилювання
elF2 .
- м’язову атрофію спричиняють пухлинний протеоліз-індукуючий фактор
та TNF-α, ангіотензин ІІ, глюкокортикоїди.
На ці ланки патогенезу кахексії загалом спрямовані сучасні пошуки методів
лікування (таргетна терапія).
92.Механізми природного протипухлинного захисту: імунні та не імунні. Сучасні наукові досягнення у імунотерапії онкозахворювань.
Імуностимулююча терапія при злоякісних новоутвореннях в даний час займає важливе місце. Головний об'єкт її дії - Т-лімфоцити, які здійснюють контроль за появою атипових клітин. Синтезовані препарати тимозин, тпмалін, тиморин, Т-активін самі по собі не мають протипухлинної дії, але вони посилюють і відновлюють діяльність ослаблених хіміотерапією або опроміненням Тклітин. Крім того, отримані певні позитивні результати активної специфічної імунотерапії (протипухлинна вакцинація). Препарати рослинного походження (фітогемаглютинін), гормон Тлімфоциту перетворюють нейтральні лімфоцити в клітини-кілери, синтез яких при злоякісних пухлинах порушений. В даний час для лікування злоякісних пухлин застосовують генну інженерію, а також антисенстерапію; принцип дії останньої полягає в зупинці функціонування конкретного гена за рахунок інгібіції синтезу відповідного протеїну. Для ефективної фармакотерапії злоякісних новоутворень необхідно діагностувати рівень пошкодження генома клітин і їх епігеномний рівень, системи клітинних сигналів.
93.Експериментальне вивчення етіології і патогенезу пухлин: методи індукції, трансплантації, експлантації. Патофізіологічні основи профілактики і лікування пухлин.
Персіваль Потт описав професійне злоякісне захворювання — рак шкіри мошонки у
сажотрусів. Спроби відтворити подібну пухлину в експерименті
закінчувалися невдачею. Учені Ішикава і Ямагіва вперше змогли викликати пухлину
у тварин. Впродовж 6 міс. вони змащували шкіру кроликів кам'яновугільною смолою
і лише після цього у тварин розвинувся рак шкіри. Індукція пухлини вірусами.
Еллерман і Банг вперше викликали лейкоз у курей за допомогою безклітинного
фільтрату з лейкозних лейкоцитів.
Індукція пухлини фізичними чинниками. Пухлину вдається відтворити за допомогою
іонізуючої радіації, у тому числі рентгенівських променів, радіоактивних
ізотопів, а також ультрафіолетових променів. Експлантація пухлини. Вирощування
пухлини вкультурі тканини поза організмом. Цей метод успішно застосовував проф.
А. Д. Тімофєєвський. Культура тканини, отримана безпосередньо з пухлини тварини
або людини, називається первинною. Даний метод особливо цінний тим, що дозволяє
вивчати індукцію пухлин і пухиногенних вірусів на людських тканинах. Пассовані
або індуковані в культурі тканини пухлинні клітини при підсадці здоровій
тварині ростуть в її організмі і утворюють злоякісну пухлину. Трансплантація
пухлини. Вперше вітчизняний вчений М. А. Новінський успішно трансплантував
пухлину дорослого собаки щенятам. Даним дослідом було покладено початок
експериментальної онкології. Метод трансплантації широко використовується і в
наш час. Є штами пассованих пухлин з добре вивченими властивостями. Алогенна
трансплантація пухлин буває успішною, тоді як така ж трансплантація здорових
тканин без імунодепресії не вдається.
Вонкології використовуються три методи лікування: хірургічний, променевий і хіміотерапевтичний.
Хірургічний метод є найбільш ефективним на ранніх стадіях пухлинного росту, і, особливо, при доброякісних пухлинах.
Променева терапія – головний метод лікування багатьох форм злоякісних новоутворень, оскільки за її допомогою вдається гальмувати ріст і, навіть, приводити до загибелі молодих клітин, які діляться. Основу механізму лікувальної дії променів на пухлинні клітини складає утворення вільних активних радикалів, перекисів, вторинних радіотоксинів, які пошкоджують клітинні мембрани, молекули ДНК і РНК, мітохондрії, в результаті чого пухлинна клітина гине за декілька днів.
Хіміотерапія включає протипухлинні препарати і гормони. Механізм дії багатьох протипухлинних препаратів полягає в порушенні синтезу нуклеїнових кислот, блокаді ферментів, порушенні біохімічних процесів, що сприяє або затримці мітозів пухлинних клітин (цитостатична дія), або до їх руйнування (цитотоксична дія).
Вивчення складних молекулярно-біологічних процесів регуляції поділу клітин, ідентифікація онкогенів, дешифрування факторів росту і їх ролі в розвитку пухлинного процесу стали основою для нового напрямку в лікуванні хворих на рак, яке отримало назву біотерапія. Біотерапія – це лікування онкологічних хворих шляхом активації природніх захисних механізмів, або введення природних полімерних молекул (цитокіни, фактори росту, та ін.). Цей напрямок дуже перспективний, так як на відміну від хіміотерапії, в якій застосовують клітинні отрути, біотерапія використовує природні для організму речовини і активує природні механізми імунного захисту. Цей вид лікування є найбільш патогенетично обґрунтованим
94.Експериментальне відтворення пухлин. Значення канцерогенних факторів у виникненні пухлин дитячого віку (на прикладі раку шкіри, щитоподібної залози, лейкозів).
Експериментально відтворити пухлину та вивчити її довго не вдавалося. Саме тому відтворення в експерименті цього патологічного процесу на початку ХХ ст. стало великим науковим досягненням. Нарешті стало можливим відкривати причини, вивчати патогенез пухлинного процесу, розробляти методи його профілактики та лікування.
Розроблені такі методи експериментального відтворення, як індукція (хімічними речовинами, вірусами, фізичними факторами); експлантація (вирощування пухлини у культурі тканини поза організмом); трансплантація (прищеплення пухлини від однієї тварини іншій).
Патогенез
Канцерогенез – тривалий процес накопичення генетичних пошкоджень. Латентний період (час від початкових змін в клітині до перших клінічних проявів) може тривати до 10-20 років. Виникнення пухлини – це багатостадійний процес, який включає 3 етапи (стадії).
І. Трансформація (ініціація) – набуття вихідною нормальною клітиною основної властивості пухлинної клітини – здатності безмежно розмножуватись і передавати цю властивість дочірнім клітинам у спадок. Усі теорії які історично підготували базу для відкриття молекулярних механізмів канцерогенезу, виходили з загальних даних, що перетворення нормальної клітини в пухлинну є результатом стійких змін в геномі клітини – мутації одного з генів, які регулюють клітинне розмноження. Внаслідок цього клітина стає ініційованою (потенційно здатною до безмежного розмноження), але потребує для проявлення цієї здатності низки додаткових умов. Факторами, які ініціюють клітину є різні канцерогени, які чинять пошкодження ДНК.
Які ж сучасні уявлення про молекулярні механізми канцерогенезу? На сьогодні встановлено, що в нормальних клітинах в ДНК є ділянка, гомологічна за нуклеотидним складом онкогену вірусів, а точніше для кожного з 20 відомих ретровірусних онкогенів в геномі нормальних і пухлинних клітин різних видів тварин є свій клітинний аналог. В нормальних клітинах клітинний аналог вірусного онкогену неактивний і має назву – протоонкоген. В клітинах пухлин він активний і має назву клітинного онкогену.
Перехід неактивного клітинного онкогену в активний клітинний онкоген проходить під впливом хімічних, фізичних і біологічних канцерогенів.
Говорячи про трансформацію нормальних клітин в пухлинні, слід зупинитися на гіпотезі Х’югса, яка у певній мірі відповідає на запитання, яким чином пухлинна клітина стає «безсмертною», тобто втрачає ліміт Хейфлика (Хейфликом встановлено, що в ядрі кожної клітини закладений генетичний механізм, який обмежує кількість мітозів клітини, наприклад, фібробласт дає 50 мітозів, а потім гине; інші клітини дають ще менше поділів і гинуть) і набуває здатність до постійного поділу, тобто ділиться тисячі і мільйони разів. Згідно цієї гіпотези, регуляція поділу в кожній клітині здійснюється системою, яка складається з трьох регуляторних генів:
1. Ген-ініціатор клітинного поділу, який кодує синтез білку-ініціатора клітинного поділу.
2. Ген-репресор І, який кодує синтез білку-репресора І. Репресор І виключає функціонування гена-ініціатора клітинного поділу.
3. Ген-репресор ІІ, який кодує синтез білку-репресора ІІ. Репресор ІІ виключає функціонування гена-репресора І.
При активації гену-репресора І синтезується репресор І, який виключає ген-ініціатор клітинного поділу, в результаті цього припиняється синтез білка-ініціатора клітинного поділу, і поділ клітин припиняється. В свою чергу, ген-репресор І знаходиться під контролем гену-репресора ІІ, який кодує синтез репресора ІІ, а він гальмує ген-репресор І. І далі, компоненти білка-ініціатора клітинного поділу здатні виключати ген-репресор ІІ. Таким чином, система регуляції клітинного поділу працює за принципом зворотнього зв’язку, що забезпечує їй автономність та певну інтенсивність клітинного поділу. «Зворотній зв’язок» в роботі системи генів, які регулюють клітинний поділ, полягає в репресії гену-репресора ІІ компонентами ініціатора клітинного поділу.
При пошкодженні гена-репресора І (вплив радіації або хімічних канцерогенів) білок-репресор І не синтезується, а тому, ген-ініціатор клітинного поділу весь час продукує ініціатор клітинного поділу – в результаті відзначається постійний безмежний поділ пухлинних клітин. Це так званий мутаційний канцерогенез.
Деякі канцерогенні фактори, наприклад, віруси, можуть створювати стійке порушення нормальної регуляції генома соматичної клітини «господаря» шляхом інтеграції з геном-репресором ІІ цієї клітини. В результаті цього ініціатор клітинного поділу може виключити тільки ген-репресор ІІ «господаря», а на вірусному гені, який інтегрований поруч з геном-репресором ІІ в клітину «господаря», буде продовжуватися синтез репресора ІІ – в кінці-кінців буде відбуватися безмежний поділ пухлинних клітин. Такий канцерогенез має назву епігеномний (геном клітини «господаря» не піддається мутації!).
ІІ. Промоція (активація). Трансформовані клітини можуть залишатися у тканині тривалий час в неактивній формі. Додатковий вплив коканцерогенного фактору, який сам не визиває трансформацію, але стимулює клітини до розмноження, приводить до того, що пухлинні клітини, які знаходяться у латентному стані, починають розмножуватися, утворюючи пухлинний вузол.
Більшість канцерогенів є повними тобто ті, які викликають і трансформацію й активацію. Механізм активації полягає в тому, що при втраті трансформованою клітиною репресора клітинного поділу або пригніченні його, для початку поділу потрібний додатковий стимул.
ІІІ. Прогресія – стійкі якісні зміни властивостей пухлини, в міру її росту, переважно у вигляді малігнізації (переродження).
Прогресія пухлини виникає під дією наступних факторів:
1. У первинний канцерогенез, як правило, втягується не одна клітина, а декілька, що сприяє формуванню в пухлині, яка розвивається, декілька субліній клітин. У пухлині яка росте, під впливом змінюючих умов (харчування, кровопостачання, інервація) її росту завжди здійснюється відбір найбільш життєздатних клітин. Певні клітини отримують перевагу. При рості пухлинної тканини в організмі змінюється гормональна регуляція. Можливе утворення антитіл проти клітин, які є в якій-небудь сублінії. В результаті, через деякий час отримує перевагу будь-яка сублінія пухлинних клітин, яка на початку складала меншість.
2. Змінагенотипу і фенотипу клітин, яка приводить до прогресії, може бути зв’язана з продовженням дії на геном пухлинних клітин канцерогенного фактору.
3. Спонтанні мутації пухлинних клітин при зниженні в них активності репаративних ферментів.
4. Набуття пухлинними клітинами нових властивостей, пов’язаних із суперінфекцією онкогенними та неонкогенними вірусами, які знаходилися в пухлинних клітинах.
Прогресія приводить до збільшення швидкості росту пухлини. Під час хіміотерапії пухлини спостерігається відбір клітин, стійких до дії лікарських засобів.
У процесі канцерогенезу і прогресії клітини втрачають своє диференціювання, вертаючись до ембріонального стану. Це явище називається анаплазією. Ознаки анаплазії, як зазначалося вище, існують в біохімічних процесах пухлинних клітин, у їх фізико-хімічних властивостях, у будові та функції. Відбувається також метаплазія – перетворення у нові клітинні форми.
Злоякісна пухлина може рости з будь-якої тканини:
меланобластома (пігментна пухлина із “родимок”), саркома (із сполучної
тканини), карцинома (із залозистої тканини)
Злоякісні
пухлини в дитячому віці мають переважно сполучнотканинну природу
95.Особливості пухлин дитячого віку: розповсюдженість, поширені типи, гістологічне походження. Поняття про ембріональні пухлини.
Пухлини у дітей. Особливості:
- часто виникають із ембріональних тканин внаслідок порушення їх розвитку та
формування – це дизонтогенетичні або тератоїдні пухлини (тератоми); -
доброякісні пухлини (ангіоми, невуси) виникають частіше, ніж злоякісні; -
саркоми (лімфосаркоми, остеосаркоми) зустрічаються частіше, ніж раки, котрі
виникають переважно у внутрішніх органах, ендокринних залозах; - злоякісні
пухлини (ембріональні нефроми, гепатоми) у дітей тривалий час зберігають
експансивний ріст, довго не метастазують і навіть здатні реверсувати –
перетворюватися у доброякісні пухлини – нейробластома у гангліоневрому; -
злоякісні пухлини у дітей частіше зустрічаються у віці 3-5 років, що засвідчує
значення внутрішньоутробних канцерогенних впливів; - деякі доброякісні пухлини
мають схильність до інфільтративного росту – ангіоми.
Класифікація: -перший тип – це дизонтогенетичні, тератоїдні пухлини або тератоми. Вони можуть бути гістіоїдні, органоїдні, організмоїдні та ембріональні, які можуть бути зрілими – тератоми та незрілими – тератобластоми. Гістіоїдні тератоми ще називають гамартомами (ангіоми, невуси, ембріональні пухлини внутрішніх органів) або гамартобластомами;
-другий тип – це пухлини з ембріональних камбіальних тканин у нервовій тканині, симпатичних гангліях, наднирниках (медулобластоми, ретінобластоми, нейробластоми). Їх також можна віднести до гамартобластом;
- третій тип – пухлини, які виникають за типом пухлин дорослих – це пухлини мезенхімального походження: гемобластоми, остеогенні та пухлини м¢яких тканин.
Дизонтогенетичні пухлини: - гамартоми та гамартобластоми судинного походження, серед яких частіше зустрічаються капілярна та кавернозна гемангіоми на шкірі (у вигляді вузлика червоно-синього кольору) та в печінці і інших органах. Капілярні гемангіоми мають здатність до інфільтративного росту, тому можуть рецидивувати після видалення. Рідше зустрічаються ангіосаркоми та лімфангіоми, які на шиї можуть досягати досить великих розмірів з проліферацією ендотелію та капілярів і інфільтративним ростом;
- гамартоми та гамартобластоми поперечно посмугованих м¢язів – рабдоміоми, які зустрічаються в серці, м¢язах кінцівок у вигляді вузла 10-15см сіро-коричневого кольору, рабдоміобластома або ембріональна рабдоміосаркома – злоякісна пухлина, яка зустрічається в органах малого тазу;
- гамартоми внутрішніх органів: нефробластома або ембріональна нефрома (пухлина Вільмса, аденосаркома) довго росте експансивно в капсулі, може досягати гігантських розмірів, рожево-білого кольору з крововиливами. Гістологічно в пухлині серед структур ниркової тканини знаходять елементи мезенхімального походження; гепатобластома або ембріональна гепатома – злоякісна пухлина печінки, на розрізі має вигляд численних біло-жовтих вузлів із солідних полів ембріональної печінкової тканини та структур мезенхімального походження. Дає метастази, ускладнюється внутрішніми кровотечами.
Тератоми і тератобластоми: тератоми організмоїдні та органоїдні – пухлини похідні з трьох зародкових листків зустрічаються в яєчках, яєчниках, середостінні, позаочеревинному просторі, основі мозку, крижово-куприковій області. В яєчниках у дівчаток частіше розвиваються злоякісні тератобластоми, в яєчках – доброякісні тератоми, тератоми зіва ростуть у вигляді поліпів, мають доброякісний перебіг, внутрішньочерепні тератоми частіше мають злоякісний перебіг, часто мають гормональну активність.
Пухлини з камбіальних ембріональних тканин: медулобластома – злоякісна пухлина в мозочку, ретинобластома – злоякісна пухлина з ембріональних недиференційованих клітин сітківки, нейробластома – злоякісна пухлина в симпатичних гангліях, мозковій речовині наднирників, швидко дає метастази, виділяє катехоламіни.
Пухлини, що розвиваються за типом у дорослих – це пухлини нервової системи: астроцитоми; кровотворної системи: лейкози, злоякісні лімфоми; пухлини кісток: остеоми, хондроми, остеосаркоми, саркоми Юінга.
96. Особливості етіологічних чинників розвитку пухлин дитячого віку. Етіопатогенезретинобластоми.
Найчастіше у новонароджених та дітей раннього віку зустрічаються дитячі гемангіоми, за даними різних авторів поширеність їх становить від 0,3% до 10%. Більшість гемангіом візуалізуються відразу після народження, частина діагностується протягом першого місяця життя. Ці доброякісні пухлини зумовлені ендотеліальною гіперплазією, яка в своєму розвитку проходить три стадії: проліферації, інволюції та атрофії.
Піококові гранульоми не бувають природженими, частіше з'являються у дітей середнього та старшого віку. Це грануляційна тканина з великою кількістю судин, появу якої інколи пов'язують з інфікуванням у разі незначної травми, але істинна причина виникнення цих утворів остаточно не з'ясована.
Рабдоміома- це доброякісна пухлина скелетних м'язів, яка також може вражати піхву. Зустрічається у жінок середнього віку.
Рабдоміома серця - доброякісна пухлина, яка зазвичай виникає в межах міокарду, пов'язана з туберозним склерозом.
97. Голодування: визначення поняття, класифікація. Зовнішні та внутрішні причини голодування. Характеристика порушень основного обміну і обміну речовин у різні періоди повного голодування (з водою).
Голодування - це стан, що виникає тоді, коли організм зовсім не одержує харчових речовин або одержує недостатню кількість їх, або не засвоює їх внаслідок хвороби.
За походженням: фізіологічне і патологічне.
Розрізняють: повне, неповне(кільк. недоїд.),часткове(якісне) голодування.
Періоди:
· 1.посил.витрачання вуглеводів; посил. катаболізму білків і гліконеогенезу; зниж ОО; негативний азотистий баланс.
· 2.переважне окиснення жирів;активізація обміну в жировій тканині;наростання концентрації кетонових тіл у крові;негазовий ацидоз.(ОО зниж.).
· 3.посилення розпаду білків життєво важливих органів;затримання води; розлад ферментних систем.
Зовнішні причини — відсутність їжі. Внутрішні причини — вади розвитку в дітей, захворювання органів травної системи, інфекційні процеси, анорексія (патологічна відсутність апетиту).
Безпосередньою причиною, що зумовлює порушення обміну при голодуванні, є розлад ферментних систем. Виникає дискоординація в процесах обміну, чітко узгоджених у нормі. Можливе накопичення проміжних токсичних продуктів. Теплопродукція підтримується протягом усього періоду голодування на мінімальному рівні і знижується до кінця третього періоду. Тепловіддача трохи скорочується. Інші життєві функції організму протягом першого і другого періоду голодування також зберігаються в межах, близьких до фізіологічних. Повне голодування без води відбувається так само, як і з вживанням води, але тяжче і недовго (3–6 днів). Якщо вода не надходить ззовні, вона береться із тканин — це оксидаційна вода. Неповне голодування (недоїдання) виникає частіше, ніж повне. Неповне - буває в тих випадках, коли організм хронічно недоотримує з їжею необхідну для енергетичних витрат кількість енергії (напр. замість 10 454–10 476 кДж (2500–3500 ккал) одержує 6280–8374 кДж (1500–2000 ккал) і менше). У зв’язку з тим, що таке голодування триває довго, розвиваються пристосувальні механізми. Основний обмін знижується більше, ніж при повному голодуванні (на 30–35% проти 10–20%). Дихальний коефіцієнт незначно знижується. Організм надзвичайно ощадливо витрачає енергетичні ресурси. Повільно зменшується маса тіла, що іноді маскується затримкою води.
98. Патофізіологія неповного та часткового (якісного) голодування. Види, причини та механізми найбільш важливих проявів. Поняття про лікувальне голодування. Наслідки голодування у дитячому віці.
Неповнеголодуваннявиникає за умов, якщоорганізмвпродовжтривалого часу не отримує з їжеюналежнукількістьенергії.Наслідкомнедоїданняможуть бути зниженняфункційтравнихорганів, ендокринноїсистеми, пам’яті, уваги і іншихпоказниківрозумовоїздатності. Оскількитакеголодуваннямаєхронічний характер, то розвиваютьсяпристосувально-компенсаторнімеханізми. Так, основнийобмінзнижуєтьсянабагатобільше – на 30-35%. Масатілазменшуєтьсяповільніше, ніж при повномуголодуванні, за рахунокзатримки води в організмі. Внаслідокзменшеннявмістубілків в кровізнижуєтьсяонкотичнийтиск, що приводить до розвиткунабряків. Виникаєгідремія, а такожанемія, брадикардія, зниженняартеріальноготиску, послаблюєтьсядихання, пригнічуєтьсястатевийінстинкт. Знижуєтьсядихальнийкоефіцієнт.При неповномуголодуваннівиникаютьнайтяжчідеструктивнізміни в тканинах, ніж при повному, оскількивонотриваєдовше. Відновитижиттєдіяльність систем організмупіслянеповногоголодування є набагатоважче.
Частковеголодуванняспостерігається при недостатньомунадходженні з їжею одного абокількоххарчовихкомпонентів (білків, жирів, вуглеводів, вітамінів) за умов нормальноїенергетичноїцінностіїжі. Часто неповнеголодуваннякомбінується з частковим.
Лікувальнеголодуванняназиваютьще методом розвантажувально-дієтичноїтерапії. Метод лікувальногоголодуваннявикористовується в лікуваннібагатьохзахворювань: серцево-судинних, шлунково-кишкових, суглобових, органівдихання, алергічних, ожиріння, цукровогодіабету другого типу, функціональнихпорушеньнервовоїсистеми, шкірнихзахворювань, ряду психічнихрозладів.Лікувальне голодування направлене на мобілізацію захисних сил організму, воно заставляє організм включати резервні сили і направити їх на нормалізацію обміну речовин, покращення травлення, кровообігу, а також очищення організму від кінцевих продуктів обміну.
У дитячому віці недоїдання, а тим паче голодування можуть призвести до необоротних наслідків. Раціон дитини має бути збалансованим за своїм складом і калорійністю.
99. Білково-калорійна недостатність, форми. Патогенез основних клінічних проявів. Квашиоркор: аліментарна білкова недостатність дитячого віку: причини, механізми, прояви.
Тривале недоїдання з переважною нестачею в складі їжі білків призводить до білково-енергетичної недостатності.
Форми: аліментарного маразму і квашіоркору (“червоний хлопчик”).
Клінічні прояви: - аліментарного маразму: знижений рівень глюкози, холестерину, нейтрального жиру в крові. Гіпопротеїнемія призводить до набряків, розвивається асцит, анемія, брадикардія, гіпотонія, порушена секреторна і моторна діяльність травного каналу. Поступово розвивається дистрофія органів і тканин. З боку нервової системи: паркінсонізм, зниження памяті. Порушення синтезу гормонів виражено в різних ендокринопатіях. Виявляються симптоми мікседеми, гіпофізарної кахексії, гіпогонадизму. Знижена стійкість проти інфекційних захворювань. Діти відстають у рості і психічному розвитку, в них розвивається депігментація шкіри, волосся, м’язове виснаження, гепатомегалія, набряки.
Квашіоркор – порушення ороговіння та інтенсивне злущування епідермісу. У дітей розвивається жирова інфільтрація печінки. Підшлункова залоза піддається гіалінозу й фіброзу, внаслідок чого знижується утворення травних ферментів, інколи розвивається цукровий діабет. Дистрофічні зміни поширюються на нирки, серце.
100. Гіпоксія: визначення поняття, класифікація, етіологія, патогенез. Обмін речовин та пристосувально-компенсаторні реакції при гіпоксії. Механізми гіпоксічного ушкодження клітин.
Гіпоксія - типовий патологічний процес, який розвивається внаслідок недостатнього забезпечення організму киснем або порушене його використання, через що знижуєтьсяенергоутворення.
Розрізняють гіпоксію місцеву та загальну.
За перебігом-гостру і хронічну.
За етіологією і патогенезом -гіпоксична, дихальна,циркуляторна,гемічна, тканинна, змішана.Компенсаторно-пристостосувальні реакції:гіпервентиляція,посилення кровообігу, збільшення кількості еритроцитів і гемоглобіну, зміни кривої дисоціації оксигемоглобіну, реакція регуляторних систем, система утилізації кисню,енергоутворення, гіпертрофія,гіперплазія,посилення еритропоезу в кістковому мозку. Патологічні реакції: зниження енергоутворення ,енергетичне голодуваннятканин,активація ПОЛ,ушкодження мембран,метаболічні зміни і накопичення продуктів неповного окиснення,ацидоз, порушення в органах і фізіологічних системах.
101. Етіологія і патогенез екзогенної гіпоксічної гіпоксії. Гірська та висотна хвороба. Зміни газового складу та рН крові.
Екзогенна гіпоксична гіпоксія - внаслідок зменшення
парціального тиску кисню у повітрі (приклад – гірська хвороба, робота в шахтах,
несправність скафандрів на глибині).
Патогенетична основа – артеріальнагіпоксемія (зменшення тиску кисню в
артеріальній крові) ->недостатнє насичення Hb киснем ->зниження
загального вмісту Hb в крові.
Механізм розвитку: підняття на висоту -> зменшення парціального тиску кисню
в повітрі -> зниження парц.тискукисню та СО2 в крові -> подразнення
хеморецепторів->гіпервентиляція
Висотна хвороба - це гостра або блискавична форма гіпоксичної гіпоксії, що
виникає під час висотних польотів у літальних апаратах з кабінами відкритого типу
або при порушенні герметичності кабін закритого
типу.
Гірська хвороба - виникає при підніманні неадаптованого організму в гори, це
приклад підгострої і хронічної гіпоксії.
При підйомі в гори з врахуванням ознак гіпоксії, виділяють наступні зони:
1) нейтральна зона (висота до 2000 м над рівнем моря). Функції організму не страждають.
2) зона повної компенсації (висота від 2000 до 4000 м над рівнем моря). Збільшення частоти пульсу і дихання, підвищення АТ, зменшується фізична і розумова працездатність, розвивається ейфорія, порушується тонка координація рухів, послабляється увага.
3) зона неповної компенсації (висота від 4000 до 6000 м над рівнем моря). Розвиваються зворотні зміни: тахікардія змінюється брадикардією, падає АТ, дихання стає частим і поверхневим, іноді розвивається дихання Чейна-Стокса, сонливість, млявість, нудота.
4) критична зона (понад 7000 м над рівнем моря). Незворотні зміни: АТ падає до 0, пульс стає ниткоподібним, з’являється термінальне дихання, людина непритомніє, розвиваються судоми і настає смерть.
Газовий склад, рН при гіпоксичній гіпоксії: зменшення р02 і рС02 артеріальної
крові, розвиток газового алкалозу.
102. Етіологія і патогенез дихальної (респіраторної) гіпоксії. Зміни газового складу та рН крові.
Респіраторний (дихальний) тип гіпоксії - виникає в
результаті:
а) недостатності газообміну в легенях у зв’язку з альвеолярною гіповентиляцією,
б) порушеннями вентиляційно-перфузійних відносин,
в) надлишковим поза- і внутрішньолегеневим шунтуванням венозної крові,
г) утруднення дифузії кисню в легенях.
Патогенетична основа- артеріальна гіпоксемія, яка часто поєднується з гіперкапнією.
Патогенез: зниженнявмісту оксигемоглобіну ->
підвищення концентрації відновленого Hb->гіперкапнія,
газовий ацидоз.
Зміни крові: зменшення р02 і збільшення рС02 артеріальної крові, розвиток
газового ацидозу.
103. Класифікація, етіологія і патогенез циркуляторної гіпоксії. Зміни газового складу та рН крові.
Циркуляторна гіпоксія — це кисневе голодування, причиною якого є розлади
загальної гемодинаміки або порушення місцевого кровообігу.
В основі порушень системного кровообігу можуть лежати недостатність серця й
недостатність судин (шок, колапс).
До місцевої гіпоксії призводять ішемія, тромбоз, емболія, венозна гіперемія.
Залежно від механізмів розвитку деякі автори виділяють дві форми циркуляторної
гіпоксії: ішемічну і застійну.
Патогенез:___________________________
Зміни крові: збільшення артеріовенозної різниці за киснем за рахунок повнішого
вилучення
його з артеріальної крові.
104. Крива дисоціації оксигемоглобіну – причини та патогенетичне значення порушень насичення гемоглобіну киснем (роль патологічних форм гемоглобіну – HbS, HbH, HbBart). Ефект Бора.
Крива дисоціації оксигемоглобіну відображує залежність між напругою кисню в
артеріальній крові і насиченням гемоглобіну киснем.
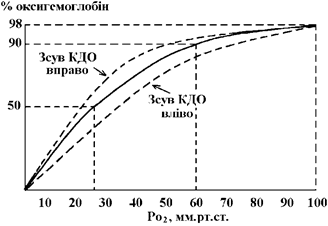
Зміщення вліво: зниження температури; алкалоз; гіпокапнія; зменшення в
еритроцитах вмісту 2,3-дифосфогліцерату; отруєння оксидом вуглецю (II); поява
спадково обумовлених патологічних форм гемоглобіну, які не віддають кисень
тканинам.
Зміщення вправо: підвищення температури; ацидоз; гіперкапнія; збільшення вмісту
в еритроцитах 2,3-дифосфогліцерату
Патологічні форми Hb:HbS — мутантний гемоглобін, у β-ланцюгах якого
в положенні 6 замість залишку глутамінової кислоти присутній залишок валіну,
яка супроводжується зниженням його спорідненості до O2 та здатністю окремих
молекул гемоглобіну злипатися між собою з утворенням ниткоподібних агрегатів.
Такі міжмолекулярні агрегати змінюють форму еритроцитів, надаючи їм
характерного серпоподібного вигляду — «серпоподібноклітинна анемія». HbBart
– складається з чотирьох гамма глобулінів, нерозчинний, накопичується в
еритроцитах, не може постачати кисень через дуже високу спорідненість.
Знаходиться в організмі при альфа-таласемії. HbH - відновлений
гемоглобін або дезоксігемоглобін, який віддав кисень тканинам(HbH – не патологічна форма, а фізіологічна).
Ефект Бора - вплив ацидозу і гіперкапнії на дисоціацію оксигемоглобіну
(приєднання кисню до дезоксигемоглобіну – вивільнення протонів, що зв’язуються
з бікарбонат-іонами, переводять їх в вуглецеву кислоту –
карбоангідразакаталізує перетворення в вуглекислий газ).
105. Етіологія і патогенез кров’яної (гемічної) гіпоксії. Патологічні форми
гемоглобіну. Зміни газового складу крові. Отруєння чадним газом та
метгемоглобінутворювачами – патогенез, прояви, профілактика.
Кров 'яна (гемічна) гіпоксія — це кисневе голодування, що виникає внаслідок
зменшення кисневої ємності крові.
Форми кров'яної гіпоксії:
а) анемічна — виникає як наслідок анемії;
б) гіпоксія, пов'язана з інактивацією гемоглобіну.
Форми інактивованогоHb:
1) Карбоксигемоглобін - продукт взаємодії гемоглобіну з оксидом вуглецю (II)
(чадним газом, CO).
2) Метгемоглобін — гемоглобін, у якому залізо перебуває в окисненому, тривалентному
стані.
3)Сульфгемоглобін — сполука гемоглобіну із
сірководнем
Зміни крові: зменшення кисневої ємності крові
Чадний газ – це кров’яна отрута, токсична дія якої пов’язана з порушенням
транспортування кисню і вираженою гіпоксією тканин, шляхом утворення в крові
карбоксигемоглобіну.
Легка форма: головний біль, запаморочення, в’ялість, шум у вухах, порушення
координації рухів, нудота, інколи блювота, сухий кашель, сльозотеча, біль в
грудях. В крові 20-30 % карбоксигемоглобіну.
Середня: короткочасна втрата свідомості, різка слабкість, загальмованість,
задишка, тахікардія, гіперемія обличчя, судоми.
Тяжка форма: коматозний стан розширення зіниць,
маятникоподібні рухи очних яблук, клонічно-тонічні судоми, підвищення
сухожильних рефлексів, поява патологічних рефлексів, можливі паралічі, дихання
типу Чейн-Стокса, розвиток гострої серцево-судинної недостатності. В крові
50-60 % карбоксигемоглобіну.
Патогенез:
а) інактивація гемоглобіну, що зменшує кисневу ємність крові, —кров 'яна
гіпоксія;
б) зміщення кривої дисоціації оксигемоглобіну;
в) зв'язування оксиду вуглецю (II) із залізом інших білків, що містять у собі гем, зокрема
цитохромів — розвивається тканинна гіпоксія.
Профілактика: при наявності пічки/каміну завжди відкривати заслінку, перевіряти
газові прилади на справність, не залишати машину з увімкненим двигуном.
106. Етіологія і патогенез тканинноїгіпоксії. Причини порушенняутилізаціїкисню. Змінигазового складу та рН крові.
Етіологія ТГ: - порушення утилізації кисню клітинами
Патогенез ТГ: 1) токсини мікроорганізмів, адреналін, тироксин у високих дозах→витікання протонів→зниження активності біологічного окислення→активаціяПОЛ→розсіювання енергії у вигляді тепла→дефіцит АТФ→роз’єднання окислення і фосфорилювання→порушенняутилізації кисню клітинами;
2) Іони важких металів→ушкодження функціональних груп дихальних ферментів→пригнічення біологічного окислення→порушення утилізації кисню
Причини порушення утилізації кисню: пригнічення біологічного окиснення, роз’єднання окиснення і фосфорилювання.
Зміни газового складу крові: зменшення артеріовенозної різниці за киснем, збільшення рО2 венозної крові.рН крові зміщується в кислу сторону
107. Порушення в систе мах організму при гіпоксії. Компенсаторно-пристосувальніреакції, значенняекспресії HIF-1 – генетично-запрограмованоївідповіді на гіпоксію.
Порушення в системах: нервова система – ейфорія, при тривалій гіпоксії -гальмування, порушення рефлекторної дiяльності, регуляцiї дихання i кровообiгу. Втрата свiдомостi i судороги є грiзними симптомами тяжкого перебiгукисневого голодування. Травна система - пригнiченняpyxoвoї функції травного каналу, зниження секреції шлунка, кишок i пiдшлункової залози. Дихальна система - розлад легеневої вентиляції, зміна ритму дихання, яке набуває часто характеру дихання Чейна-Стокса, розвиток застiйних явищ у легенях:потовщення альвеолярно-капiлярної мембрани, розвиток в нiйфiброзної тканини, погiршеннядифузiї кисню з альвеолярного повiтря у кров.Ендокринна система: у кopi надниркових залоз первинна активiзацiязмiнюється виснаженням функції.Видільна система: первинна полiурiязмiнюється порушенням фiльтрацiйноїздатностi нирок.
У тяжких випадках гiпоксії знижується температура тiла,що пояснюється зниженням обмiну речовин i порушенням терморегуляції.
ПротеїнHIF-1 посилює синтез білків, які відповідають за адаптацію до кисневого голодування. Серед них: ферменти біологічного окислення, білки, що входять до складу дихальних ланцюгів мітохондрій і системи окисного фосфорилювання, ферменти гліколізу, судинний ендотеліальний фактор росту. Також HIF-1 затримує процеси диференціації клітин, що біологічно виправдане в умовах гіпоксії. Вилучення генів, які кодують структуру HIF-1 з геному призводить до внутрішньоутробної загибелі плода.
Компенсаторно-пристосувальні реакції
1) гіперплазія кісткового мозку та посилення еритроцитопоезу; 2) гіпертрофія мітохондрій(збільшення кількості дихальних ланцюгів → відновлюється або збільшується здатність клітини виробляти енергію всупереч нестачі кисню в крові, що притікає); 3) гіпертрофія дихальних м’язів та міокарда(внаслідок посилення кровопостачання цих органів за рахунок утворення нових капілярів під дією факторів росту→ збільшення маси легеневих артеріол, кількості міоглобіну в м’язах, гіпертрофія кіркової речовини наднирників, гіпоталамусу, нейронів дихального центру); 4) Метаболічні пристосувальні реакції (посилення глюконеогенезу, активація безкисневого утворення енергії шляхом гліколізу, активація гексозофосфатного шунта для синтезу сполук для відновлення НАДФ/НАДФН, посилення синтезу ферментів антиоксидантного захисту, простагландинів,);
108. Системні та місцеві механізми екстреної адаптації к гіпоксії. Механізми довготривалої адаптації до хронічній гіпоксії.
Системні:реакції зовнішнього дихання, реакції системного кровообігу (збільшення кровопостачання тканин за рахунок збільшення ХОК, перерозподілу крові, підвищення артеріального тиску), реакції системи крові (збільшення кисневої ємності крові за рахунок збільшення кількості еритроцитів та концентрації гемоглобіну в периферичній крові, а також зміщення кривої дисоціації оксигемоглобіну вправо).
Місцеві: 1) Тканинні реакції, спрямовані на покращення забезпечення клітин киснем (артеріальна гіперемія, ангіогенез, підвищення кількості міоглобіну в м’язах); 2) Реакції в системі утилізації кисню (активація гліколізу, збільшення спорідненості цитохромоксидази з киснем, підвищення ступеня спряженості окиснення й фосфорилювання, збільшення кільскості дихальних ферментів і мітохондрій, зниження функціональної активності клітин→зменшення потреби клітин у кисні )
Механізми довготривалої адаптації: зниження основного обміну та потреб у кисні, посилення газообмінної функції легень, збільшення рівня еритроцитів і гемоглобіну у крові, економія реакцій регуляторних систем і органів на додаткові стимули →підвищення стійкості клітин до гіпоксії
109. Патофізіологічні основи профілактики та лікування гіпоксічних станів. Гіпербарична оксигенація (ГБО). Можливі негативні наслідки кисневої терапії.
У випадках, коли утилізація кисню в клітинах не порушена, застосовують замісну терапію, антиоксиданти та антигіпоксанти. Однак ефект лікарських засобів залежить від виду та стадії гіпоксії, а при порушення мікросомального окислення в печінці змінюється і їх фармакодинаміка. В профілактиці гіпоксичних станів знижують чутливість до дефіциту кисню за допомогою моделювання станів, які супроводжуються гальмуванням центральної нервової системи та зниженням обміну речовин (наркоз, гіпотермія) та підвищенням стійкості до гіпоксії за допомогою тренувань в умовах барокамери або високогір’я.
Гіпербарична оксигенація – метод кисневої терапії, який передбачає дихання в умовах високого атмосферного тиску у барокамерах. Мета даного методу – збільшити кількість фізично розчиненого в плазмі крові кисню. Цей вид кисневої терапії показаний при всіх видах гіпоксії,(отруєння барбітуратами, СО, вроджені вади серця) окрім тканинної.
Негативні наслідки кисневої терапії –гіпероксія, порушення кальцієвого гемостазу, утворення активних форм кисню, утворення ПОЛ, підвищення проникності плазматичних мембран для кальцію, його транспорту з позаклітинної рідини в клітину і потім в мітохондрії.
110. Механізми більш високої стійкості новонароджених до гіпоксії порівняно з дорослими.
Новороджені діти більш стійкі до гіпоксії завдяки наявності фетального гемоглобіну та ізоформ ферментів.Рефлекторні механізми для регуляції дихання новонароджених малят людини ще не сформовані так, як у дорослих, проте їх дихальний центр може функціонувати в умовах браку кисню до 20 хв. Високі значення компенсаторних та адаптаційних показників новонароджених в умовах гіпоксії порівняно з дорослими організмами спостерігаються завдяки активації у ниханаеробного глікогенезу та більш високій здатності тканин до утилізації кисню.
111. Порушення енергетичного обміну: етіологія, патогенез, наслідки. Поняття про енергетичні потреби організму, позитивний та негативний енергетичний баланс. Експериментальне визначення основного обміну. Зміни основного обміну при патології.
Залежно від причин розвитку розрізняють поза- і внутрішньо клітинні розлади енергетичного обміну.
Позаклітинниминазиваютьрозладиенергетичногообміну, пов'язані з первиннимипорушеннямизабезпеченняклітинпоживнимиречовинами і киснем. Це голодування і більшістьвидівгіпоксії.
При внутрішньоклітиннихрозладахенергетичногообміну доставка поживнихречовин і кисню не страждає, а порушуєтьсяїхвикористанняклітинами.
Основні причини:
1. Повна відсутність їжі або дефіцит поживних речовин у ній.
2. Порушення процесів травлення і всмоктування.
3. Порушення мобілізації поживних речовин з депо (наприклад, ураження печінки, розлади нервово-гуморальної регуляції жирового обміну).
4. Порушення транспорту поживних речовин кров'ю (загальні й місцеві розлади кровообігу).
5. Порушення дифузії поживних речовин у тканинах.
6. Втрата поживних речовин або їх використання не за призначенням (протеїнурія, глюкозурія, опікова хвороба, злоякісні пухлини).
Дефіцит АТФ у клітині призводить до:
1. Порушення механічної роботи - скорочення контрактильних структур клітин. Це виявляє себе розладами елементарних клітинних функцій: скорочення, міграції, екзо- і ендоцитозу, клітинного поділу, руху війок, джгутиків (рис. 59).
2. Порушення осмотичної роботи — процесів активного транспорту іонів. При
дефіциті АТФ страждають механізми як первинного, так і вторинного активного транспорту: натрій-калієвий і кальцієвий насоси, натрій-кальцієвий і натрій- водневий обмінні механізми. Це викликає порушення внутрішньоклітинного гомеостазу й ушкодження клітин.
3. Порушення хімічної роботи — біосинтезу речовин. Наслідком цього є порушення самовідновлення й самовідтворення клітин, механізмів їх довгострокової адаптації до дії факторів навколишнього середовища. В остаточному підсумку відбувається повільна загибель клітин.
4. Порушення реакцій клітинного метаболізму.
Енергетичний баланс — співвідношення енергії, що надходить до організму з їжею, та енергії, що витрачається внаслідок діяльності організму.
Основний обмін — це мінімальні енергетичні витрати організму, визначені ранком у стані повного спокою, натще (через 12-14 год. після останнього прийому їжі), в умовах температурного комфорту (t = 18-20 °С, вологість повітря - 60-80 %, швидкість його руху - 0,1-0,2 м/с).
Зовнішні фактори:
а) кліматичні умови;
б) час доби;
в) висота над рівнем моря;
г) характер харчування; ґ) характер виробничої діяльності.
Внутрішні фактори:
а) ріст і маса тіла;
б) площа поверхні тіла;
в) вік;
г) стать..
Збільшення основного обміну характерне для гіперфункції щитоподібної залози, аденогіпофіза, статевих залоз.
Зменшення основного обміну реєструють при гіпофункції щитоподібної залози, аденогіпофіза, статевих залоз; при хронічній недостатності кори надниркових залоз, при голодуванні.
112. Характеристика порушень вуглеводного обміну; критерії еуглікемії, гіпоглікемії, гіперглікемії, порушень толерантності до глюкози. Роль змін нейро-гуморальної регуляції вуглеводного обміну в патогенезі гіпо- і гіперглікемічних станів.
Основнв причини порушень вуглеводного обміну.
1. Порушення перетравлювання вуглеводів та їх усмоктування в кишках ).
2. Порушення вуглеводної функції печінки.
3. Порушення катаболізму глюкози в периферичних клітинах.
4. Порушення нейрогормональної регуляції вуглеводного обміну.
Гормони, що беруть участь у регуляції вуглеводного обміну, поділяють на дві групи: інсулін і контрінсулярні гормони.
Контрінсулярними називають гормони, які за своїми біологічними ефектами є антагоністами інсуліну. До них належать адреналін, глюкагон, глюкокортикоїди, соматотропний гормон.
Гіпоглікемія - це зменшення концентрації глюкози в плазмі крові до рівня, який обумовлює появу клінічних симптомів, що зникають після нормалізації вмісту цієї речовини.
Ознаки гіпоглікемії виникають, як правило, при зменшенні вмісту глюкози нижче 4 ммоль/л.
Гіперглікемія — це збільшення вмісту глюкози в плазмі крові (понад 6,66 ммоль/л при визначенні методом Хагедорна - Йєнсена).
Механізми:
1. Збільшення надходження глюкози в кров. Це буває після приймання їжі (аліментарна гіперглікемія), при посиленні глікогенолізу й глюконеогенезу в печінці (зменшення вмісту інсуліну або збільшення концентрації контрінсулярних гормонів).
2. Порушення використання глюкози периферичними тканинами.
Так, при зменшенні вмісту інсуліну порушується надходження й утилізація глюкози в інсулінозалежних тканинах (м'язах, жировій тканині, печінці)
Цукровий діабет - це хвороба, яка у нелікованому стані виявляє себе хронічним збільшенням вмісту глюкози в крові - гіперглікемією (визначення ВООЗ, 1987).
1. Панкреатичний цукровий діабет- видалення в собак 9/10 підшлункової залози (Мерінг і Мінковський, 1889).
2. Алоксановий цукровий діабет: однократне введення тваринам алоксану — речовини, що вибірково ушкоджує Β-клітини острівців підшлункової залози.
3. Стрептозотоциновий цукровий діабет: введення тваринам антибіотика - стреп-
тозотоцину, що вибірково ушкоджує )3-клітини острівців.
4. Дитизоновий цукровий діабет: введення тваринам дитизону — речовини, що
зв'язує цинк і в такий спосіб порушує депонування й секрецію інсуліну.
5. Імунний цукровий діабет - введення тваринам антитіл проти інсуліну.
6. Метагіпофізарний цукровий діабет — тривале введення тваринам гормонів
адено-гіпофіза - соматотропного гормону, АКТГ.
7. Метастероїдиий цукровий діабет — тривале введення тваринам
глюкокортикоїдів.
8. Генетичні моделі цукрового діабету — виведення чистих ліній мишей та інших
тварин зі спадково обумовленою формою хвороби.
113. Класифікація, причини і механізми розвитку гіпоглікемічних станів. Патогенез та прояви гіпоглікемічної коми. Профілактика та невідкладні заходи при гіпоглікемії у дітей.
Гіпоглікемія - це зменшення концентрації глюкози в плазмі крові до рівня, який обумовлює появу клінічних симптомів, що зникають після нормалізації вмісту цієї речовини.
Ознаки гіпоглікемії виникають, як правило, при зменшенні вмісту глюкози нижче 4 ммоль/л.
Механіхми:
1. Зменшення надходження глюкози в кров. Це буває при голодуванні, порушеннях травлення (недостатність амілолітичних ферментів, розлади всмоктування), при спадкових і набутих порушеннях глікогенолізу і глюконеогенезу в печінці.
2. Посилене використання глюкози на енергетичні потреби організму (наприклад, важка фізична робота).
3. Втрата глюкози (глюкозурія) або використання її не за призначенням (злоякісні пухлини).
КЛІНІЧНІ ознаки:
Клінічні ознаки гіпоглікемії пов'язані з двома групами порушень в організмі.
I. Порушення постачання глюкозою тканин головного мозку. Залежно від ступеня гіпоглікемії розвиваються такі симптоми, як головний біль, неможливість зосередитися, стомлюваність, неадекватна поведінка, галюцинації, суцоми, гіпоглікемічна кома.
II. Активація симпатоадреналової системи. Цим обумовлені серцебиття, посилене потовиділення, тремтіння, почуття голоду.
Гіпоглікемічна кома є найтяжчим проявом гіпоглікемії, і якщо вчасно не надати допомогу (введення глюкози), призводить до смерті. Вона характеризується втратою свідомості, зникненням рефлексів, порушеннями життєво важливих
функцій
Лікування гіпоглікемічних станів.
При підтвердженні діагнозу гіпоглікемічної коми вводять 50 мл 50% розчину глюкози. В Україні доступним є 40% розчин глюкози, який вводять внутрішньовенно в дозі 40–60 мл. У деяких хворих відновлення свідомості відбувається швидко, «на кінці голки», в інших — займає певний проміжок часу. Якщо ефект сумнівний, додатково вводять 100 мл гідрокортизону та за відсутності протипоказань (ІХС, гіпертонічна хвороба) — 1 мл 0,1% розчину адреналіну, який сприяє мобілізації глікогену печінкою з подальшим підвищенням рівня глюкози у крові.
Останнім часом рекомендують введення 1 мг глюкагону внутрішньом’язово. Гіперглікемічний ефект препарату зумовлений його глікогенолітичною дією, тому він неефективний при виснаженні глікогену в печінці, наприклад, у разі голодування, гіпокортицизму, печінкової недостатності [1].
Хворі без свідомості підлягають терміновій госпіталізації. Їм необхідний моніторинг глікемії. Продовжується внутрішньовенне введення глюкози протягом усього часу очікуваної дії інсуліну чи перорального цукрознижувального препарату, які викликали цю кому. Наприклад, якщо кома викликана прийомом хлорпропаміду, введення глюкози слід проводити протягом кількох днів [3].
У деяких випадках корисно додавати до програми лікування:
бікарбонат натрію для помірного залуження сечі, що скорочує період напіввиведення більшості цукрознижувальних засобів;
окреотид по 50–100 мкг двічі на добу, який знижує секрецію інсуліну.
Якщо свідомість не відновилася, потрібно повторно визначити рівень глікемії та провести обстеження, спрямоване на виключення інших причин коми. Подальша інтенсивна терапія включає: штучну вентиляцію легень, інфузійну терапію та глюкокортикоїди.
Для інфузійної терапії застосовують розчини глюкози (розчин ГіК), розчини гідроксиетильованих крохмалів (гекодез) та похідні багатоатомних спиртів (реосорбілакт, ксилат). Крім впливу на об’єм циркулюючої крові останні дві групи препаратів під час метаболізму частково перетворюються на глюкозу і підвищують рівень глікемії, що є безумовно бажаним ефектом у хворих з гіпоглікемією.
Слід також зазначити, що у частини пацієнтів з гіпоглікемією можливий розвиток кетоацидозу — так званого кетоацидозу голодування. Таким пацієнтам у програмі інфузійної терапії потрібно передбачити введення препаратів антикетогенної дії, наприклад ксилату (5–6 мл/кг на добу протягом 1–2 днів). Патогенетично обґрунтованим і перспективним є використання у цих пацієнтів препарату глюксил, основними діючими складовими частинами якого є глюкоза, ксилітол та натрію ацетат.
Профілактика гіпоглікемічних станів.
Найпростіший профілактичний захід, який потрібно рекомендувати кожному хворому, — постійно мати при собі шматочки цукру та при появі перших ознак гіпоглікемії приймати їх по 10–20 г. Важливе значення має також регулярний контроль рівня глікемії крові, особливо в періоди змін режиму харчування та при значних фізичних навантаженнях. В цілому, будь-який епізод гіпоглікемії можна певною мірою розглядати як результат помилки лікаря або пацієнта або як наслідок неадекватності схеми інсулінотерапії. Необхідно обов’язково знайти причину розвитку гіпоглікемії та оцінити вірогідність розвитку її рецидивування. По можливості, слід направити пацієнта в школу діабету та порадити носити з собою картку із зазначеним діагнозом та описом заходів, які слід провести при розвитку гіпоглікемії. Пацієнт має постійно пам’ятати про ризик розвитку гіпоглікемії; лікар повинен нагадувати йому про це на кожній консультації.
114.Класифікація та етіопатогенез синдрому неонатальної гіпоглікемії
Неонетальна гіпоглікемія проявляється одразу ж після народження або в перші 12-72 часи життя в 1-3 із 1000 новонароджених. У доношених новонароджених симптоми гіпоглікемії розвиваються за рівня глюкози - 1,7 ммоль/л, у недоношених – 1,1 ммоль/л. Спостерігається найчастіше у дітей, які народилися з масою, меншою від 2,5 кг, недоношених, із внутрішньоутробною затримкою розвитку, за порушення актів смоктання та ковтання, гемолітичної хвороби гіалінових мембран, за наявності гіпотрофії та вродженої патології вуглеводного обміну. Транзиторна гіпоглікемія спостерігається у 80-90% дітей, народжений від матерів, які мали діабет вагітних або хворіють на ЦД. У 10-20% дітей із групи високого ризику розвивається стійка тяжка гіпоглікемія. Тяжка та тривала неонатальна гіпоглікемія призводить до ураження головного мозку дитини. Неонатальна гіпоглікемія в подальшому може стати причиною значного відставання у психомоторному розвитку дитини. Повторні епізоди гіпоглікемії призводять до органічного ураження ЦНС (пірамідні, екстра пірамідні та мозочкові порушення). Безпосередніми причинами гіпоглікемії у новонароджених є гіперінсулінемія, спадковий дефект ферментів метаболізму вуглеводів (наприклад, глюкозо-6-фосфатази), дефіцит речовин – джерел глюкози (наприклад, глікогену), дефіцит контрінсулярних гормонів. Не діагностовано та не лікована гіпоглікемія призводить до незворотних уражень нервової системи, епілептичних нападів, психічних розладів, відставання у розумовому розвитку. Надто це стосується тяжких, тривалих і частих гіпоглікемій.
Ранніми ознаками гіпоглікемії у новонароджених є м’язова гіпотонія, тремор ціаноз, порушення дихання, судоми, сонливість, апное, фасцикуляції, зниження температури тіла. Важливими диференційно-діагностичними ознаками у цей період є низький рівень глюкози на тлі клінічних симптомів, зникнення симптомів після нормалізації рівня глюкози, виникнення симптомів внаслідок зниження рівня глюкози (тріада Уіппла). Транзиторна гіпоглікемія може мати безсимптомний перебіг.
У разі підтвердження наявності гіпоглікемії невідкладно проводять в/в інфузію глюкози зі швидкістю 6-8 мг/кг/хв.. (максимальний об’єм – 80 мл/кг/добу), дозу глюкози зменшують поступово. Бажано залишати дитину на грудному годуванні. Запобігання гіперглікемії у вагітної, хворої на діабет (рівень глюкози в крові має не перевищувати 11,0 ммоль/л), знижує ризик розвитку гіпоглікемії у новонародженого, що у свою чергу зменшує ризик виникнення макросомії, дихальних розладів, синдрому Жильбера (гіпербілірубінемії новонароджених), гіпокальціємії, вроджених вад розвитку.
Моніторинг рівня глюкози, тактика спостереження та лікування гіпоглікемії у недоношених дітей залишаються дискусійними. Останні експериментальні дослідження свідчать, що епізоди гіпоглікемії, що повторюються, справляють шкідливий вплив на розвиток мозку, пригнічують розвиток клітин і викликають апоптоз у головному мозку. Вважають за необхідне проводити обов’язкове визначення рівня глюкози у недоношених дітей і починати активне лікування, якщо рівень глюкози нижчий від 2,5 ммоль/л.
Прогноз у випадку транзиторної гіпоглікемії більш сприятливий, іноді ймовірні мінімальні порушення інтелекту. У випадку прогресування або рецидиву гіпоглікемії необхідно виключити наявність у дитини гіперісулінемії, дефіциту контрінсулярних гормонів (ГР, кортизолу, глюкагону) або вроджених порушень, глюконеогенезу чи синтезу глюкагону. Гіпоглікемія, обумовлена цими порушеннями, зазвичай виявляється у віці 3-6 міс. (збільшення перерви між годуванням до 8 годин). Проводять пробу з глюкагоном (30 мкг/кг/в/в або в/м – після введення глюкагону концентрація глюкози в плазмі зростає більш наж на 2,2 ммоль/л). Характерниими ознаками дефіциту ГР є мікропенія та вади обличчя по середній лінії; глюкогенозу – гепатомегалія; гіперінсулінемії – макросомія, відсутність або низький вміст кетонових тіл у сечі, низький рівень жирних кислот у крові. Діагноз гіперінсулінемії підтверджується, якщо на тлі гіпоглікемії рівень інсуліну у сироватці перевищує 72 ммоль/л.
115. Визначення поняття, класифікація цукрового діабету (ВООЗ). Загальна характеристика основних типів цукрового діабету (тип інсулінової недостатності, її походження, особливості перебігу, типові прояви, ускладнення і патогенетичні принципи лікування).
Цукровий діабет - це хвороба, яка у нелікованому стані виявляє себе хронічним збільшенням вмісту глюкози в крові - гіперглікемією (визначення ВООЗ)
1. Панкреатичний цукровий діабет- видалення в собак 9/10 підшлункової залози (Мерінг і Мінковський, 1889).
2. Алоксановий цукровий діабет: однократне введення тваринам алоксану — речовини, що вибірково ушкоджує Β-клітини острівців підшлункової залози.
3. Стрептозотоциновий цукровий діабет: введення тваринам антибіотика - стреп-
тозотоцину, що вибірково ушкоджує )3-клітини острівців.
4. Дитизоновий цукровий діабет: введення тваринам дитизону — речовини, що
зв'язує цинк і в такий спосіб порушує депонування й секрецію інсуліну.
5. Імунний цукровий діабет - введення тваринам антитіл проти інсуліну.
6. Метагіпофізарний цукровий діабет — тривале введення тваринам гормонів
адено-гіпофіза - соматотропного гормону, АКТГ.
7. Метастероїдиий цукровий діабет — тривале введення тваринам
глюкокортикоїдів.
8. Генетичні моделі цукрового діабету — виведення чистих ліній мишей та інших
тварин зі спадково обумовленою формою хвороби.
Цукровий діабет І типу — інсулінозалежшй. Він характеризується абсолютною інсуліновою недостатністю, що виникає в результаті загибелі β-клітин панкреатичних острівців. Розвивається в осіб молодого віку, зазвичай до 30 років. Тому його ще називають ювенільним, або юнацьким. Супроводжується кетозом — накопиченням кетонових тіл. Необхідне лікування інсуліном.
Цукровий діабет II типу — інсулінонезолежний. Він характеризується відносною недостатністю інсуліну або інсулінорезистентністю. Розвивається у дорослих, найчастіше після 40 років. Не супроводжується кетозом. У його лікуванні інсулін не застосовують.
Цукровий діабет І типу — захворювання з генетичною схильністю, у виникненні якого важливу роль відіграють фактори зовнішнього середовища (рис. 63). Спадкова схильність до діабету І типу обумовлена антигенасоційованим характером цього захворювання. Так, доведений зв'язок між виникненням цукрового діабету І типу і наявністю певних HLA-генів (В8, Bwl5, Dw3, Dw4) у головному комплексі гістосумісності (МНС). Це наводить на думку про значення аутоімунних механізмів у розвитку цієї хвороби.
Серед факторів зовнішнього середовища, здатних ушкоджувати β-клітини панкреатичних острівців, велике значення мають р-цитотропні віруси (віруси Коксакі,
епідемічного паротиту, кору) і fi-цитотропні хімічні агенти (наприклад, алоксан, стрептозотоцин).
Причини розвитку цукрового діабету ІІ типу.
1. Спадкова схильність. Вона, однак, на відміну від діабету І типу, не пов'язана з генами головного комплексу гістосумісності (МНС).
2. Ожиріння. Його відзначають у 80 % хворих. Тому виділяють дві форми цукрового діабету II типу:
а) з ожирінням;
б) без ожиріння.
На відміну від діабету І типу фактори зовнішнього середовища не мають особливого значення в етіології діабету II типу.
Патогенез цукрового діабету II типу з ожирінням. Умовно виділяють два етапи патогенезу. І. Гіперінсулінемічний етап. Споживання великої кількості їжі особами з ожирінням викликає збільшення секреції інсуліну (гіперінсулінемія). Ця реакція спрямована на активацію процесів депонування поживних речовин у жировій тканині.
Гіперінсулінемічний етап патогенезу цукрового діабету II типу з ожирінням
М'язи ж не мають потреби в дії інсуліну. Тому вони оберігають себе від надлишку цього гормону зменшенням кількості рецепторів на поверхні м'язових клітин. Розвивається явище інсулінорезистентності м'язової тканини — її чутливість до дії інсуліну падає. II. Гіпоїнсулінемічний етап (рис. 66). Підвищене навантаження на інсулярний апарат може призводити до функціонального виснаження β-клітин. Цьому сприяють генетично обумовлені їхні дефекти і надлишок в організмі контрінсулярних гормонів. Як наслідок, секреція інсуліну падає й розвивається його відносна недостатність. При цьому дія інсуліну на жирову тканину зберігається (на жирових клітинах багато рецепторів до інсуліну), а на м'язову тканину - зменшується внаслідок інсулінорезистентності.
Клінічно це виявляє себе розвитком гіперглікемії (немає дії інсуліну на м'язову тканину) і відсутністю кетозу (зберігається дія інсуліну на жирову тканину).
Ускладнення. Макроангюпатії, мікроангюпатії, нейропатії.
Основні патогенетичні принципи лікування цукрового діабету.
1. Уведення інсуліну при інсулінозалежному цукровому діабеті І типу. Перспективними в цьому плані є трансплантація (3-клітин острівців підшлункової залози й застосування автоматизованих систем дозування і введення інсуліну.
2. Уведення фармакологічних препаратів, що усувають гіперглікемію, - гіпоглікемічних засобів (бігуаніди, похідні сульфонілсечовини).
3. Дієтотерапія, що забороняє вживати харчові продукти з високим вмістом цукру, регулює енергетичну цінність їжі та режим її споживання.
4. Фізичні навантаження (тренування). Вони зменшують рівень гіперглікемії й збільшують чутливість м'язової тканини до інсуліну.
116. Етіологія цукрового діабету 1-го типу (значення спадкових факторів та факторів середовища в розвитку абсолютної інсулінової недостатності). Патогенез цукрового діабету 1 типу: порушення білкового, вуглеводного, жирового, водно-електролітного обмінів і кислотно-основного стану. Клінічні прояви.
Цукровий діабет І типу - захворювання з генетичною схильністю, у виникненні якого важливу роль відіграють фактори зовнішнього середовища Серед факторів зовнішнього середовища, здатних ушкоджувати В-клітини панкреатичних острівців, велике значення мають В-цитотропні віруси (віруси Коксакі,епідемічного паротиту, кору) і В-цитотропні хімічні агенти (наприклад, алоксан, стрептозотоцин).Генетична схильність, пов’язана з генами МНС, виявляє себе, з одного боку, утворенням аутоантитіл, здатних вибірково ушкоджувати В-клітини острівців підшлункової залози, з другого - зменшенням резистентності клітин до дії екзогенних ушкоджувальних факторів (В-цитотропних вірусів і хімічних агентів). Усе це разом призводить до ушкодження інсулярного апарату й загибелі В-клітин. Утворення інсуліну припиняється.
Порушення білкового обміну проявляються:
1. Аміноацидемісю - збільшенням вмісту амінокислот у плазмі крові В основі цього лежить зменшення транспорту амінокислот у м’язові клітини і посилення протеолізу в м’язах, унаслідок чого вивільнені амінокислотинадходять у кров.
2. Порушеннями біосинтезу білків. Це прямо пов’язано з випадінням анаболічної дії інсуліну.Клінічно виявляє себе порушеннями фізичного й розумового розвитку дітей, уповільненням загоєння ран, порушеннями утворення антитіл, унаслідок чого збільшується чутливість до інфекцій, часто розвивається фурункульоз.
Порушення вуглеводного обміну
1. Збільшення надходження, глюкози в кров із печінки. Знімається гальмівний вплив інсуліну на ферменти глікогенолізу й глюконеогенезу,унаслідок чого збільшується інтенсивність цих процесів у печінці.
2. Зменшення використання глюкози інсулінозалежними тканинами. Це пов’язане з тим, що при дефіциті інсуліну:
а) зменшується проникність клітинних мембран для глюкози у м’язовійі жировій тканині;
б) зменшується утворення глікогену в печінці і м’язах;
в) падає активність пентозного циклу в печінці й жировій тканині;
г) зменшується активність гліколізу в усіх інсулінозалсжних тканинах;
ґ) відбувається пригнічення ферментів циклу Кребса в печінці і м’язах;
д) порушується перетворення глюкози в жири у печінці й жировій клітковині.
Все це призводить до гіперглікемії.
Порушення жирового обміну
1.Гіперліпацидемія - пов’язана з активацією ліполізу і пригніченням ліпогенезу в жировій тканині внаслідок порушення балансу між інсуліном і контрінсулярними гормонами
2. Кетоз (гіперкетонемія й кетонурія). Не використаний Ацетил-КоА у синтезі жирних кислот і у циклі Кребса є джерелом посиленого кетогенезу.
3. Гіперліпопротеїнемія. Збільшенням вмісту ліпопротеїдів дуже низької щільності (ЛПДНЩ)
4. Жирова інфільтрація печінки. Є наслідком надмірного надходження в печінку вільних жирових кислот. Якщо можливості гепатоцитівутворювати міцели ЛПДНГ вичерпуються, надлишок тригліцеридів відкладається в печінкових клітинах.
5. Схуднення. Гальмуванняліпогенезу при відсутності інсуліну й пригнічення реакцій гліколізу.Посилення ліполізу під дією контрінсулярнихгормонів.
6. Атеросклероз. Пов’язаний з гіперглікемією, гіперліпідемією та ацидозом.
Порушення водно-електролітного обміну
1. Зневоднення (дегідратація). Є наслідком поліурії.
2. Гіпернатріємія (через активацію ренін-ангіотензиновоїистеми)
Порушення кислотно-основного стану:
Негазовий ацидоз. Залежно від механізмів його розвитку виділяють:
а) кетонемічний метаболічний ацидоз - пов’язаний з накопиченням кетонових тіл;
б) лактацидемічний метаболічний ацидоз - пов’язаний з накопиченням молочної кислоти(внаслідок гіпоксії при гіповолемії).
Клінічні прояви діабету: поліурія (осмотичний діурез внаслідок глюкозурії), полідипсія (посилена спрага), ознаки дегідратації, які, переважно, є помірно вираженими (зниження еластичності шкіри, сухість шкіри та слизових оболонок), спричинені зневодненням слабкість та сонливість, схуднення (рідше), кетоацидоз та кетоацидотична кома (іноді перший помічений симптом), схильність до гнійних інфекцій шкіри чи інфекцій сечостатевої системи.
117. Поняття про діабетичну ембріо- та фетопатію. Їхні патогенез та наслідки для дитини.
Цукровий діабет жінки негативно впливає на розвиток плоду, особливо в жінок з лабільним перебігом захворювання при частих епізодах гіпоглікемії та кетоацидозу.Формуються два симптомокомплекси — діабетична ембріопатія та фетопатія.
Механізм дії. При недостатньо компенсованому або латентному діабеті глюкоза з крові матері трансплацентарно поступає до плоду, викликаючи гіперплазію В-клітин підшлункової залози та гіперінсулінізм, сприяючи таким чином збільшенню внутрішніх органів та надлишкової маси тіла. Значно зменшується швидкість міграції та диференціації клітин, наступають дегенеративні зміни в нервових та гліальних клітинах.
До діабетичної ембріопатії відносять вади розвитку, які формуються в ембріональному періоді, найчастіше — це вади розвитку кістковом’язової, серцево-судинної системі, хвостового відділу хребта, вертебральнадисплазія, аненцефалія.
Діабетична фетопатія (ДФ) - це відхилення у розвитку плоду, які виникають після першого триместру вагітності при погано компенсованому або латентному діабеті у матері. Проявляється наступними ознаками: кушингоїдний тип, порушення функції життєво важливих систем організму дитини, метаболічні зміни, затримка процесів адаптації в ранньому неонатальному періоді.
Наслідки. В новонароджених з діабетичною фетопатією найчастіше зустрічаються такі вроджені вади розвитку, як аномалії скелету (37%), аномалії кардіоваскулярної системи (24%), аномалії нервової системи (14%).Кожна четверта дитина з діабетичною фетопатією страждає на кардіомегаліюта порушення серцевого ритму.Як вже зазначалося, такі діти страждають на макросомію, ацидоз, гіпоглікемію, порушення електролітного балансу та синдром дихальних розладів через недорозвиненість легенів. Одразу після пологів можуть виникати неонатальні кровотечі та неонатальнатромбоемболія. Рівень перинатальної смертності в таких випадках становить 26%.
118. Етіологія та патогенез діабетичного кетоацидозу у дітей, хворих на цукровий діабет.
Діабетичний кетоацидоз (ДКА) — це гостре, небезпечне для життя ускладнення цукрового діабету, яке характеризується розвитком гіперглікемії, кетоацидозу та кетонурії.
Етіологія.Основними факторами високого ризику виникнення ДКА у дітей унаслідок абсолютного дефіциту інсуліну є:
• маніфестація ЦД;
• поганий метаболічний контроль або епізоди ДКА в минулому;
• у дівчаток – перипубертатний вік;
• психічні порушення, у тому числі з розладами режиму харчування;
• складні або нестабільні відносини в сім'ї (наприклад, жорстоке поводження
батьків);
• ненавмисні або свідомі пропуски ін'єкцій інсуліну.
• обмежений доступ до медичної допомоги.
• введення неякісного інсуліну
• приєднання інфекційних захворювань, що викликають блювання чи діарею.
• також у відповідь на стресс
Патогенез
Оскільки при діабеті порушується розщеплення глюкози, вмикаються алтернативні шляхи добування енергії – розщеплення жирів, їх посилений метаболізм у печінці супроводжується продукцією надмірної кількості проміжних і кінцевих метаболітів. Кетонемія та кетонурія призводять до розвитку метаболічного ацидозу зі значним зниженням рівнів рН і бікарбонатів у сироватці крові. Глюкозурія, характерна для діабетичного ацидозу, призводить до осмотичного діурезу, дегідратації та гіперосмолярності плазми крові. Внаслідок декомпенсованої дегідратації розвивається преренальна ниркова недостатність. Гіперглікемія, осмотичний діурез, гіперосмолярність сироватки крові та метаболічний ацидоз індукують важкі електролітні порушення. Гіперосмолярність сироватки крові, дегідратація та ацидоз призводять до підвищення осмолярності в клітинах мозку, що клінічно проявляється зміною рівня свідомості.
119. Патогенез клінічних проявів діабетичного кетоацидозу у дітей, хворих на цукровий діабет. Основні напрямки терапії діабетичного кетоацидозу.
Симптоми і ознаки діабетичного кетоацидозу включають симптоми гіперглікемії разом з нудотою, блювотою і, особливо у дітей, болями в животі. Більш важка декомпенсація метаболізму проявляється загальмованістю і сонливістю. У пацієнтів може бути гіпотензія та тахікардія, обумовлені зневодненням і ацидозом. В якості компенсації ацидемії розвивається часте і глибоке дихання (дихання Куссмауля). Також можливий фруктовий запах при диханні, що викликається присутністю в повітрі, що видихається ацетону. В деяких випадках розвивається набряк мозку (частіше у дітей, рідше у підлітків), Гіперосмолярність сироватки крові, дегідратація та ацидоз призводять до підвищення осмолярності в клітинах мозку, та їх набухання.Набряк мозку найчастіше спостерігається у дітей <5 років, у яких ДКА є першим проявом цукрового діабету. Групу найбільшого ризику становлять діти з високим рівнем азоту сечовини в крові і низьким Paco2. Додаткові фактори ризику - пізня корекція гіпонатріємії і застосування бікарбонатів при лікуванні діабетичного кетоацидозу.
Терапія.
1. Проведення регідратації — необхідно перелити 6–10 л розчинів в/в впродовж 24 год
2. Зменшення гіперглікемії — слід розпочати в/в інсулінотерапію (застосовується інсулін короткої дії).
3. Поповнення дефіциту калію:
4. Компенсація ацидозу:
1) ацидоз легкого та помірного ступеня компенсується в міру наводнення, введення інсуліну та вирівнювання порушень водно-електролітного балансу;
2) гідрокабонат натрію вводьте лише при pH артеріальної крові <6,9 (i лише до моменту, коли pH досягне 7,0);
5. Знайдіть причину, яка спровокувала розвиток даного стану, та розпочніть відповідне лікування (напр. антибіотикотерапію у разі інфекції).
120. Етіологія, патогенез цукрового діабету 2-го типу. Роль спадкових факторів. Причини відносноїінсулінової недостатності. Поняття інсулінорезистентності. Порушення обміну речовин і фізіологічних функцій. Клінічні прояви.
Цукровий діабет II типу - інсулінонезалежний. Він характеризується відносною недостатністю інсуліну або інсулінорезистентністю. Розвивається у дорослих, найчастіше після 40 років. Не супроводжується кетозом. У його лікуванні інсулін не застосовують.
Етіологія.
1. Спадкова схильність. Вона, однак, на відміну від діабету І типу, не пов’язана з генами головного комплексу гістосумісності (МНС).
2. Ожиріння. Його відзначають у 80 % хворих
На відміну від діабету І типу фактори зовнішнього середовища не мають особливого значення в етіології діабету II типу.
Умовно виділяють два етапи патогенезу.
І. Гіперінсулінемічний етап. Споживання великої кількості їжі особами з ожирінням викликає збільшення секреції інсуліну (<гіперінсулінемія). Ця реакція спрямована на активацію процесів депонування поживних речовин у жировій тканині.М’язи ж не мають потреби в дії інсуліну. Тому вони оберігають себе від надлишку цього гормону зменшенням кількості рецепторів на поверхні м’язових клітин. Розвивається явище інсулінорезистентності м’язової тканини - її чутливість до дії інсуліну падає.
II. Гіпоінсулінемічний етап. Підвищене навантаження на інсулярний апарат може призводити до функціонального виснаження В-клітин. Цьому сприяють генетично обумовлені їхні дефекти і надлишок в організмі контрінсулярних гормонів. Як наслідок, секреція інсуліну падає й розвивається його відносна недостатність. При цьому дія інсуліну на жирову тканину зберігається (багато рецепторів до інсуліну), а на м’язову тканину - зменшується внаслідок інсулінорезистентності. Клінічно це виявляє себе розвитком гіперглікемії (немає дії інсуліну на м’язову тканину) і відсутністю кетозу (зберігається дія інсуліну на жирову тканину).
При ЦД 2 типу спостерігаються ті ж зміни в метаболізмі, що і при 1 типі, але значно менш виражені
Клінічні прояви діабету: поліурія (осмотичний діурез внаслідок глюкозурії), полідипсія (посилена спрага), ознаки дегідратації, які, переважно, є помірно вираженими (зниження еластичності шкіри, сухість шкіри та слизових оболонок), спричинені зневодненням слабкість та сонливість, схильність до гнійних інфекцій шкіри чи інфекцій сечостатевої системи.
121. Ускладнення хронічної гіперглікемії (цукрового діабету). Причини та механізми різних видів ком при цукровому діабеті. Віддалені ускладнення діабету.
Віддалені
ускладнення цукрового діабету –
цевторинніпорушення, яківикликанінедостатністю в кровіінсуліну:
• ретинопатія – ураженнясітківки ока у
виглядімікроаневризм, точковихкрововиливів, набряку,
можезакінчитисявідшаровуваннямсітківки;
• ангіопатія – порушенняпроникностісудин,
підвищенняїхньоїламкості, схильність до тромбозу, атеросклерозу;
• енцефалопатія – змінапсихіки, настрою,
емоційналабільність, депресія;
• нефропатія – мікроальбумінурія, протеїнурія,
ниркованедостатність;
• нейропатія – порушеннятемпературної та
больовоїчутливості, оніміння, відчуттяпечіння, щопочинаються в
дистальнихвідділахкінцівок;
• діабетична стопа – ураження стоп у
виглядігнійно-некротичнихпроцесів, виразок, кістково-суглобовихуражень,
щовиникає на фонізмінипериферичнихнервів, судин, шкіри, м’яких тканин, кісток,
суглобів.
Види ком при цукровому діабеті
1. Діабетичнакетонемічеськая
кома. У основіїїрозвитку лежать ацидоз і інтоксикація,
обумовленікетоновимитілами. 2. Гіперосмолярна кома.
Розвиваєтьсяунаслідокдегідратації головного мозку,
обумовленоювисокоюміроюгіперглікемії 3. Лактацидемічеськая грудки.
Обумовленанакопиченняммолочноїкислоти і пов'язаним з цим ацидозом. 4.
Гіпоглікемічна кома. Можерозвиватися в результатіпередозуванняінсуліну при
лікуванніцукровогодіабету.
122.
Експериментальнемоделюванняцукровогодіабету. Принципипрофілактики і
терапіїйогоосновнихтипів. Профілактикаускладненьцукровогодіабету.
1. Панкреатичнийцукровийдіабет- видалення в собак 9/10 підшлунковоїзалози (Мерінг і Мінковський, 1889).
2. Алоксановийцукровийдіабет: однократневведеннятваринамалоксану — речовини, щовибірковоушкоджує Β-клітиниострівцівпідшлунковоїзалози.
3. Стрептозотоциновийцукровийдіабет: введеннятваринамантибіотика - стреп-тозотоцину, щовибірковоушкоджує )3-клітини острівців.
4. Дитизоновийцукровийдіабет: введеннятваринамдитизону — речовини, щозв'язує цинк і в такийспосібпорушуєдепонування й секреціюінсуліну.
5. Імуннийцукровийдіабет - введеннятваринамантитілпротиінсуліну.
6. Метагіпофізарнийцукровийдіабет — тривалевведеннятваринамгормонівадено-гіпофіза - соматотропного гормону, АКТГ.
7. Метастероїдиийцукровийдіабет — тривалевведеннятваринамглюкокортикоїдів.
8. Генетичнімоделіцукровогодіабету — виведеннячистихлініймишей та іншихтваринзіспадковообумовленою формою хвороби.
123. Порушенняліпідногообміну: причини,
механізми, прояви. Залежністьрозвиткудисліпопротеїнемійвідфакторівсередовища,
спадковості, супутніхзахворювань. Принципикласифікаціїдисліпопротеїнемій.
Причини розладів жирового обміну можуть бути порушення:
- Перетравлювання і всмоктування ліпідів у тонкій кишці
- Транспорту ліпідів кров’ю
- Депонування ліпідів у жировій тканині
- Жирової функції печінки
- Обміну жирів у периферичних тканинах
- Нервової та гормональної регуляції
Дисліпопротеїдемія - Група захворювань, при яких внаслідок порушень утворення, транспорту та утилізації ліпопротеїдів змінюється їх рівень та склад у плазмі крові. Включає в себе гіперліпопротеїдемії, гіполіпопротеїдемії, аліпопротеїдемії та комплексні порушення (з надлишком одних та нестачею інших ліпопротеїдів).
Сімейна дис- β -ліпопротеїнемія, або «флотуюча» гіперліпемія. Характерні: а) високий рівень холестерину і ТГ у плазмі; б) ксантелазми, ліктьові і колінні ксантоми, жовтувато-коричневі відкладення ліпідів у шкірі долонних ліній і в місцях тиску кілець; в) атеросклероз коронарних артерій, периферичних судин і судин мозку; в) ожиріння, цукровий діабет, гіпотиреоз.
124. Етіологія і патогенез первинних
(спадкових) і вториннихгіперліпопротеїнемій.
Генетичнідефекти , які можуть бути причиною розвиткупервинних (спадкових) гіперліпопротеїнемій.
1. Генетичнообумовленізміниструктуриапопротеїнів — білковоїчастиниліпопро-теїднихміцел, унаслідокчоголіпопротеїдиплазмикрові не можутьвзаємодіяти з відповідними рецепторами абозазнаватиферментативнихперетворень.
2. Спадковідефектиферментів, щоберуть участь в обмініліпопротеїдів, зокрема, дефіцитліпопротеїнліпази, печінковоїліпази, лецитинхолестеролацилтрансфера-зи (ЛХАТ).
3. Аномаліїклітиннихрецепторів до ліпопротеїдів.
Можливі причини розвиткувторинних (набутих)гіперліпопротеїнємій.
1. Ендокринніхвороби (цукровийдіабет, гіпотиреоз).
2. Метаболічнірозлади (ожиріння, подагра).
3. Хворобинирок (нефротичний синдром).
4. Хворобипечінки (обтураційнажовтяниця).
5. Інтоксикації (алкоголізм).
125. Ожиріння: визначенняпоняття,
класифікації; етіологія і патогенез окремих форм.
Експериментальнемоделюванняожиріння. Медичніпроблеми, пов’язані з ожирінням.
Первиннимназиваютьожиріння, яке є самостійнимпатологічнимпроцесом. Вториннеожиріння є ознакою тих абоіншихзахворювань. Найбільшпоширенимиваріантамивторинногоожиріння є церебральне і гормональнеожиріння. основні причини первинногоожиріння. 1. Надлишковийвжитокїжі, щоперевищуєенергетичнівитратиорганізму. 2. Гіподинамія — обмеженняфізичноїактивностілюдини. 3. Генетичнасхильність. Можевиявлятися в особливостяххарчовоїповедінкилюдиниабо в особливостяхрегуляції жирового обміну. Церебральнеожиріннявиникає при поразкахгіпоталамуса, де зосередженіцентри, регулюючіхарчовуповедінку. Такіпоразкивиникають при травмах, пухлинах, енцефаліті. Провідниммеханізмоможиріння в цьомувипадку є поліфагія (підвищенняапетиту). Гормональнеожиріннярозвивається як одна з ознакендокринних хвороб. Воносупроводжуєрозвиток: а) гіпотиреозу; б) аденомиострівцівпідшлунковоїзалози (гіперінсулінізм); у) синдрому Іценко-Кушинга; г) гіпофункціїстатевихзалоз. Гіперпластичнеожирінняпов'язане з гіперплазієюжировихкліток, тобтоіззбільшеннямїхкількості. Для ньогохарактерні початок в ранньомудитячомувіці і велика надлишкова вага. У основігіпертрофічногоожиріннялежитьзбільшеннямасиокремихжировихкліток, при цьомуїхкількість не міняється. Ожирінняцього типа маєбільшпізніше почало і не настількивиражено, як у попередньомувипадку.
I. Експериментальнімоделіпервинногоожиріння. Отриманочистілініїмишей і щурів з генетичнообумовлениможирінням, що є ознакою, яка передаєтьсявід потомства до потомства.
II. Експериментальнімоделівторинного церебрального ожиріння:
а) руйнуваннявентромедіальних ядер гіпоталамуса, щоутворюють "центр насичення"
б) електростимуляціялатеральних ядер гіпоталамуса, щостановлять "центр апетиту".
III. Експериментальнімоделі вторинного гормонального ожиріння:
а) вимиканняфункціїдеякихендокриннихзалоз (видаленнящитоподібноїзалози, кастрація);
б) введення в організмвеликоїкількостідеякихгормонів (інсуліну, глюкокор-тикоїдів).
IV. Експериментальні моделі місцевогоожиріння — перетинаннясимпатичнихнервів.
При цьому в тканині з порушеноюіннервацієюзбільшуєтьсямасажировоїклітковини,
оскількиприпиняється ліполітична діякатехоламінів.
Головний
принцип боротьби з ожирінням – створення і підтримання негативного
енергетичного балансу організму, аж доки не буде досягнуто бажаної маси тіла.
Зазначена мете передбачає комплекс лікувальних заходів у двох напрямках: 1.
Зменьшення надходження калорій в організм; 2. Збільшення використання калорій
організмом
126.Етіологія та класифікація синдромів генетичного ожиріння у дітей.
◇Етіологія:
-ураження гіпоталамічних шляхів, що беруть участь у регулюванні енергетичного балансу;
-Підвищена активність ферментів ліпогенеза;
-Сниження активності ферментів ліполіза,
-дефіцит гормону роста;
-гіпотиреоз;
-синдром Кушинша.
◇Класифікація синдромів генетичного ожиріння у дітей:
І.Ожиріння із затримкою розвитку:
1)Домінантне успадкування:
●Синдром Прадера-Віллі
●Спадкова остеодистрофія Олбрайта
●Дефіцит SIM1( в квадраті)
●Дефіцит ВDNF/ TrkB
2) Рецесивне успадкування:
●Синдром Барде-Бідля
●Дефіцит TUB( в квадраті)
ІІ. Ожиріння без затримки розвитку:
1)Домінантне успадкування:
●Синдром Альстрема
●Дефіцит MC4R(в кубі)
●Дефіцит SH2B1
●Дефіцит KSR2
2) Рецесивне успадкування:
●Дефіцит лептину
●Дефіцит рецептора лептину
●Дефіцит POMC
●Дефіцит PCSK1
127.Позитивний і негативний баланс азоту. Види гіперазотемії. Зміни білкового складу крові.Спадкові порушення обміну амінокислот.
◇Позитивний азотистий баланс – це стан, при якому кількість азоту, що надходить, переважає над кількістю виведеного азоту. Позитивний азотистий баланс – це нормальний стан для організму, що росте. Наприклад,він характерний для дітей ,які потребують більшої кількості білка для процесів росту, нормального фізичного і розумового розвитку.Також характерний для вагітних, спортсменів, в яких іде ріст м’язової маси (культуристи) і для людей у період одужання після важких інфекційних захворювань або після тривалого голодування, коли білок витрачається на відновлення тканин.
◇ Негативний азотистий баланс – це стан, при якому кількість виведеного азоту переважає над кількістю азоту, що надходить в організм. Він розвивається при білковому голодуванні, коли кількість білка на добу становить нижче 30 г.
◇Види гіперазотемій:
1) Ретенційна/ ниркова( при захворюваннях нирок).
2) Продукційна/ печінкова ( при здорових нирках).
3) Обтураційна( виникає при закупорці сечових шляхів).
◇Зміни білкового складу крові:
Вміст загального білка в плазмі при народженні
становить 47-65 г/л, у недоношених — менше 50 г/л, причому рівень білка
залежить від ступеня недоношеності. Особливо низький вміст а- та (3-
глобулінів, до складу яких входять високоспеціалізовані білки.
Концентрація загального білка та білкових фракцій у новонароджених
залежить від стану матері при вагітності. У дітей, які народилися від матерів з
різними формами пізнього токсикозу, розвивається гіпо- та диспротеїнемія,
що проявляється гіпоальбумінемією та підвищенням рівня ос,-, а2- та |3-
глобулінів.
Протягом першого року життя відбувається зниження вмісту загального
білка в сироватці крові; особливо низькі показники у дітей віком 2-6 тиж., а
починаючи з 6 міс. відбувається поступове підвищення його рівня. Проте в
молодшому шкільному віці вміст загального білка дещо нижчий, ніж в
середньому у дорослих, причому ці відхилення більш чітко виражені у
хлопчиків.
Вміст загального білка в плазмі крові дітей, старших року, збільшується
переважно за рахунок підвищення рівня альбуміну та у-глобулінів.
◇Спадкові порушення обміну амінокислот:
-аміноацидопатії
-алкаптонурія
-альбінізм
-тирозиноз
-лейциноз
-Фенілкетонурія
-Гістидинемія
-Триптофанурія
-Хвороба «кленового сиропу «
-Гомоцистинурія
-Гіпофосфатазія
-Хвороба Німана-Піка
-Хвороба Гоше
128. Порушення пуринового та піримідинового обміну. Етіологія, патогенез подагри.
◇Порушення пуринового обміну
Кінцевим продуктом обміну пуринових основ( аденіну та гуаніну), які належать до структури нуклеїнових кислот, є сечова кислота.
Порушення її синтезу і виділення призводить до гіперурикемії.
Найтяжчим варіантом первинної гіперурикемії є подагра.
Подагра-полігенна хвороба, розвиток якої залежить від дії додаткових факторів зовнішнього середовища та способу життя людини( споживання великої кількості м'яса, алкоголю, гіподинамія, геохімічні особливості території). Також роль відіграє генетична схильність до подагри визначається кількома генами, серед яких найбільше значення мають патологічні алелі ГФРТ і ФРПС.
◇Фактори ризику подагри:
-надмірне надходження пуринів в організм.
-надмірне надходження в організм молібдену.
-стать( частіше хворіють чоловіки).
-літній вік, для якого характерна вікова гіперурикемія.
-спадкова схильність.
Вторинна гіперурикемія з розвитком подагричного синдрому пов'язана переважно з порушенням виведення сечової кислоти нирками.
Хронічна ниркова недостатність, полікістоз нирок, нефропатія вагітних досить часто призводить до затримання в організмі сечової кислоти.
Зниження секреції сечової кислоти в нирках спостерігається в разі ацидозу будь-якої етіології, гіпотиреозу, свинцевого отруєння.
◇Патогенез подагри:
Ураження органів і систем під час подагри пов'язаний з відкладанням уратів, ушкодженням тканин кристалами солей сечової кислоти та розвитком запального процессу.
Найуразливіша в цьому аспекті є хрящова тканина.
Відкладання солей спричинює гостре подагричне запалення, яке супроводжується болем, гарячкою і закінчується утворенням подагричних вузлів, деформацією суглобів.
Відкладання уратів у нирках пов'язане з розвитком сечокам'яної хвороби (уролітіазу) та подагричної нефропатії.
◇Порушення обміну піримідинових основ:
Катаболізм піримідинових основ( цитозину, тиміну, урацилу) призводить до утворення Бета-амінокислот.
Найбільше значення має оротацидурія-спадкова автономно-рецесивна хвороба, пов'язана з дефіцитом двох ферментів- оротатфосфорибозилтрансферази та оротидин-5-фосфатдекарбоксилази.
При оротацидурії накопичується -оротова кислота.
Ця речовина кристалізуєтьсч з утворенням конкрементів у сечовивідних шляхах.
Найнебезпечніші зміни характерні для системи крові в разі оротацидурії.
Наприклад, порушення поділу еритробластів призводить до тяжкої мегалобластної анемії, рефрактерної до лікування ціанокобаламіном( Вітамін В12).
Також спостерігається імунодефіцит, пов'язаний з недостатнім утворенням Т-лімоцитів.
129.Спадкові порушення обміну амінокислот: фенілкетонурія, алкаптонурія, альбінізм, тирозиноз. Можливості їх фармокорекції у дитячому віці.
◇Фенілкетонурія
- захворювання зааутосомно-рецесивним типом спадкуванням.
-Його причиною є генетичний дефект ферменту фенілаланінгідроксилази, що в нормі перетворює фенілаланін на тирозин.
-В результаті цього захворювання, фенілаланін накопичується у великій кількості в крові, тканинах і спинномозковій рідині, що вперші ж місяці життя веде до важкого ураження ЦНС й невиліковного слабоумства.
◇Тирозиноз
-Виникає внаслідок генетичного дефекту ферменту-оксидази парагідроксифенілпіровиноградної кислоти.
-накопичується в крові й разом з тирозином виводиться із сечею.
◇Алкаптонурія
-є наслідком порушення синтезу оксидази гомогентизинової кислоти, що перетворює останню в малеїлацеооцтову кислоту.
-у результаті в крові й сечі з'являється гомогентизинова кислота.
-Гомогентизинова кислота з крові проникає в тканини- хрящову, сухожилля,зв'язки, внутрішній шар стінки аорти, унаслідок чого з'являються темні плями в ділянці вух, носа, на склерах.
Іноді розвиваються тяжкі зміни в суглобах.
◇Альбінізм
-обумовлений дефіцитом ферменту тирозинази.
-унаслідок цього не утворюється пігмент шкіри й волосся-меланін.
◇Можливості їх фармокорекції у дитячому віці:
На прикладі фенілкетонурії:
– обмеження надходження в організм фенілаланіну.
– дієтотерапія: зменшити кількість вживаного фенілаланіну;
– призначають білкові гідролізати (цимогран, лефанолак, берлофен, гіпофенат) або амінокислотні суміші, без фенілаланіну, які стають головними продуктами харчування, що забезпечують потребу в білку;
– ноотропніпрепарати;
– вітаміни;
130.Гіпо- та гіпервітамінози: види, причини і механізми розвитку. Патогенез основних клінічних проявів. Принципи корекції вітамінної недостатності.
Гіпервітаміноз- вид патології, що виникає при надмірному надходженні вітамінів до організму.
◇Підвищена потреба у вітамінах:
1. Особливі фізіологічні стани організму (інтенсивний ріст, вагітність, лактація).
2. Особливі кліматичні умови.
3. Інтенсивне фізичне навантаження.
4. Інтенсивне нервово-психічне навантаження, стрес.
3. Інфекційні стани та інтоксикації.
6. Дія шкідливих виробничих факторів.
7. Захворювання внутрішніх органів та залоз внутрішньої секреції.
8. Підвищена екскреція вітамінів.
◇Патогенез клінічних проявів:
-розлади обміну речиновин, травлення та недокрів'я( при гіпервітамінозі ретинолу (А));
-порушення жирового обміну, втрата маси тіла, підвищується вміст Са і Р в крові та надлишкове відкладення їх у кістках, нирках, кровоносних судинах, серці( віт. D3);
-алергійні реакції у вигляді висипів на шкірі, безсоння, кровотечі через підвищення ламкості капілярів( віт. С).
Гіповітаміноз- патологічний стан, який розвивається внаслідок зменшення вмісту певного вітаміну(або вітамінів ) в організмі.
Вітамінна недостатність супроводжується розладами біохімічних та фізіологічних процесів і виникненням специфічної патології.
◇Основні причини і класифікація:
1)зменшення надходження певного вітаміну в організм у складі продуктів харчування — екзогенні/ аліментарні гіповітамінози.
2) порушення засвоєння певних вітамінів клітинами організму (внаслідок розладів їх всмоктування у травному тракті або нездатності біохімічних систем організму включати вітамін в обмінні процеси, зокрема внаслідок наявності їх структурних конкурентів — антивітамінів) — ендогенні гіповітамінози.
3) збільшене виведення вітаміну з організму або підвищена його утилізація в біохімічних та фізіологічних процесах (в період годування грудьми, вагітності, виснажливій фізичній праці, екстремальних температурних умовах, тяжких інфекційних хворобах тощо).
◇Патогенез клінічних проявів при гіповітамінозі:
-порушення репродуктивної системи, підвищення проникності і ламкості капілярів( гіповітаміноз вітаміна Е);
-ураження нервової системи, серцево-судинної системи с розвитком аритмій( гіповітаміноз В1);
-кровоточивість ясен, випадіння зубів, зниження імунітету( гіповітаміноз С);
-розвиток мегалобластної анемії, оніміння нижніх кінцівок, слабкість, випадіння волосся, діарея і анорексія ( гіповітаміноз В12).
◇Принципи корекції вітамінної недостатності:
Профілактика та лікування вітамінної недостатності – гіпо- та авітамінозів полягає у сприянні збільшення їх кількості в організмі ентеральним шляхом з їжею і призначенням вітамінних препаратів.
Для корекції вітамінного балансу використовують лікувальну і профілактичну тактики.
•Лікувальну тактику використовують,коли доведений дефіцит якогось певного вітаміну. Вона здійснюється під контролем лікаря високими лікувальними дозами необхідних вітамінів певними курсами фармакотерапії, як правило одним препаратом (вітаміном).
•Профілактичне спрямування корекції балансу вітамінів здійснюється нижчими дозами, ближчими до добової потреби. Для попередження дефіциту вітамінів розроблені вітамінні й вітамінно-мінеральні комплекси, які призначаються курсами раз у рік по 2-6 тижнів.
131. Особливості гіпо- та гіпервітамінозів у дитячому віці(А,Д,Е,В)
Особливості обміну вітамінів у дітей: При нормальному харчуванні матері під час вагітності в організмі доношеної дитини до народження знаходиться достатній запас вітамінів, особливо А, Д, Е, В12; Після народження дитина отримує вітаміни з молозивом, потім протягом 3-4 місяців – зі зрілим материнським молоком; Активація вітамінного обміну відбувається в печінці; В зв’язку із більш інтенсивним обміном потреба у вітамінах в дітей вища, ніж у дорослої людини; Засвоєнню вітаміну А у грудних дітей сприяє ліпаза грудного молока; У недоношених дітей запас вітамінів значно менший, ніж у дорослих; У недоношених дітей гіповітаміноз розвивається через малий запас вітамінів в організмі, функціональну неповноцінність печінки та інших органів виникає надзвичайно легко!!!
Гіповітаміноз А.Особливості клініки у дітей:у дітей грудного віку — попрілість, пліснявка, стоматит, бронхіт, знижується стійкість організму до інфекцій. При тривалому дефіциті ретинолу діти відстають у масі та зрості.Профілактика полягає в правильному годуванні дітей.
Гіповітаміноз Д.Рахіт патогенез:дефіцит вітаміну Д--зменшення всмоктуваннякальцію в кишечнику--гіпокальциемія\гіпофосфатемія--порушення мінералізації кісток --гіперпаратиреоз--демінералізація і резорбція кісток.
Клінічні прояви: збудливічть,пітливість,неспокій,деформації кісток(куряча грудна клітка,нитки перлів,сплощена потилиця).
Профілактика 500МОвітаміну Д.
Гіповітаміноз Е.Особливості клініки у дітей:нестача вітаміну Е може спостерігатися у новонароджених дітей, оскількитокофероли погано проникають через плаценту. Наслідком дефіциту вітаміну Е може з'явитися гемолітична анемія, ретинопатія, порушення зору. У новонароджених дітей через Е-вітамінну недостатність може розвинутися раптова смерть. Є точка зору, що бронхопульмональна дисплазія Е наслідком дефіциту вітаміну Е у плода.Первинна недостатність вітаміну Е розвивається у немовлят при штучному вигодовуванні, особливо при надлишку поліненасичених жирних кислот, а також у дітей при нестачі білка в раціоні.
Гіповітаміноз В1:У дітей чітко виражені зміни з боку нервової системи: вони примхливі, швидко втомлюються, скаржаться на невизначені болі по ходу нервів. Сухожильні рефлекси знижені. У маленьких дітей нерідко спостерігається гіперстезія, плаксивість, поганий сон, згасання рефлексів, загальна і часткова скутість.
Гіпервітаміноз Д виникає при передозуванні ергокальциферолу або при підвищеній чутливості організму до його препаратів.
Причинами гіпервітамінозу Д: 1) неправильне шаблонне призначення дітям профілактичних і лікувальних доз ергокальциферолу без урахування клінічних умов, пори року, віку, характеру вигодовування і стану дитини; 2) передозування ергокальциферолу в 3—10 разів; 3) неправильне зберігання спиртового розчину ергокальциферолу, випаровування рідкої частини і збільшення концентрації препарату в 2—5 разів; 4) одночасне призначення ергокальциферолу і УФО; 5) підвищена чутливість до ергокальциферолу (ідіопатична гіперкальціємія) у дітей з хронічними захворюваннями нирок.
Спостерігаються раннє закриття швів і тім'ячок, задишка, підвищення артеріального тиску, глухість тонів серця, систолічний шум, тахікардія, збільшення печінки і селезінки, рідкі випорожнення або запор. Вміст фосфору в крові може бути підвищеним, нормальним або зниженим, в сечі — підвищеним. Проба Сульковича різко позитивна (гіперкальціурія). Рентгенологічне визначається відкладення кальцію в зонах росту кісток.Лікування гіпервітамінозу D залежить від тяжкості стану. Виключають продукти, що містять кальциферол і кальцій (сир, молоко). Каші краще готувати на овочевому відварі. При тяжких і середньої важкості формах проводять інтенсивну дезинтоксикаційну і регідратаційну терапію.
Гіпервітаміноз А:спостерігається в грудному віці при випадковому або помилковому введенні вітаміну в надмірній дозіі проявляється гідроцефалією , випинанням тім'ячка , лихоманкою , втратою апетиту , блювотою , підвищенням тиску ліквору.
Гіпервітаміноз Е:при внутрішньовенному введенні вітаміну Е у новонароджених може наступити раптова смерть, асцит,печінкова і ниркова недостатність, тромбоцитопенія, гіпербілірубінемія
132. Порушення водно-сольового обміну.Форми гіпо- і гіпергідрії,етіологія,патогенез.Компенсаторно-пристосувальні механізми.
Співвідношення між кількістю спожитої і виділеної води називають водним балансом. Якщо води виділяється менше, ніж надійшло в організм, то це характеризується як позитивний баланс. Він супроводжує виникнення набряків, зокрема, при значному ослабленні серцевої діяльності, голодуванні. Якщо води виділяється більше, ніж поступило в організм, виникає від’ємний баланс. Цей стан може бути наслідком порушення функції нирок, деяких захворювань гіпофізу, зокрема, при нестачі виділення антидіуретичного гормону.
Дегідратація:
1. Ізоосмолярним називають зневоднення, при якому осмотичний тиск плазми крові й міжклітинної рідини не міняється. Воно розвивається у випадках еквівалентної втрати води й електролітів. Це спостерігається іноді при поліурії, розладах діяльності кишок, а також зразу після гострої крововтрати.
2. Гіпоосмолярне зневоднення характеризується зменшенням осмотичного тиску позаклітинної рідини і виникає у випадках переважної втрати солей. Воно розвивається при втраті секретів шлунка й кишок (пронос, блювота), а також при підвищеному потовиділенні, якщо втрата води відшкодовується питвом без солі.
3. Гіперосмолярним називають зневоднення, при якому збільшується осмотичний тиск позаклітинної рідини. Це спостерігається в тих випадках, коли втрата води перевищує втрату електролітів (насамперед натрію), наприклад, при гіпервентиляції, профузному потовиділенні, втраті слини (піт і слина гіпотонічні стосовно крові), а також при проносі, блювоті й поліурії, коли відшкодування втрати води її надходженням в організм є недостатнім.
Гіпергідратація:
1.Ізоосмолярна гіпергідрії осмотичний тиск позаклітинної рідини не змінюється. Цей вид порушень може спостерігатися протягом деякого часу після введення надлишкових кількостей ізотонічних розчинів.
2. Гіпоосмолярна гіпергідрія (водне отруєння) характеризується зменшенням осмотичного тиску позаклітинної рідини. Цей вид гіпергідрії в експерименті на тваринах моделюють повторними введеннями води в шлунок, особливо на тлі
введення вазопресину, альдостерону або видалення надниркових залоз. У клініці водне отруєння можливе при рефлекторній анурії, а також у другій стадії гострої ниркової недостатності.
3. Гіперосмолярна гіпергідрія, для якої характерне збільшення осмотичного тиску позаклітинної рідини, може розвиватися при вживанні для пиття солоної морської води.
Позаклітинне зневоднення (гіпогідрія, дегідратація, ексикоз) — це зменшення об'єму позаклітинної рідини. В основі його лежить негативний водний баланс.
Причинами позаклітинного зневоднення можуть бути:
І. Недостатнє надходження води в організм:
а) екстремальні ситуації, у яких може опинитися людина під час землетрусу, у пустелі (повне водне голодування);
б) неможливість самостійно вгамувати спрагу (важкохворі люди, грудні діти);
в) порушення формування відчуття спраги при ураженнях питного центру в ЦНС.
II. Втрата води організмом:
а) виділення великої кількості рідини нирками (поліурія), наприклад, при цукровому й нецукровому діабеті;
б) втрата рідини через травний канал (нестримна блювота, проноси, гіперсалівація);
в) посилене потовиділення, наприклад, при інтенсивній фізичній роботі, при дії високої температури;
г) збільшення виділення вологи з видихуваним повітрям при всіх видах задишки; ґ) втрата води з ексудатом при запаленні (набряки, запалення серозних оболонок).
д) крововтрата.
Позаклітинне зневоднення (гіпогідрія, дегідратація, ексикоз) — це зменшення об'єму позаклітинної рідини. В основі його лежить негативний водний баланс.
Захисно-пристосувальні механізми при позаклітинному зневодненні:
1. перехід рідини з інтерстиціального сектора в судини,через зменшується гідростатичний тиск крові в капілярах, з одного боку, і збільшується онкотичний тиск крові внаслідок її згущення
2. Зменшення об'єму циркулюючої крові, пов'язане зі зневодненням, веде до збудження волюморецепторів і, в кінцевому підсумку, до збільшення секреції антидіуретичного гормону(збільшує реабсорбцію води в нирках, обмежуючи її втрату організмом)
3. Зменшення об'єму циркулюючої крові викликає активацію ренін-ангіотензинної системи і збільшення секреції альдостерону корою надниркових залоз. Це веде до збільшення реабсорбції іонів натрію в нирках і до нормалізації осмотичного тиску позаклітинної рідини.
4. У результаті зменшення артеріального тиску збуджуються барорецептори, що призводить до активації симпатоадреналової системи .
5. Зневоднення через центральні й периферичні механізми викликає відчуття спраги. У результаті формуються поведінкові реакції, спрямовані на пошук води й поповнення втраченої рідини.
Причиною внутрішньоклітинного зневоднення є збільшення осмотичного тиску міжклітинної рідини, пов'язане з розвитком гіпернатріємії.У результаті зневоднення збільшується внутрішньоклітинна концентрація електролітів, що веде до порушення гідратних оболонок білкових молекул.
Серед загальних порушень на перший план виходять розлади функції нейронів ЦНС. Це виявляється розвитком нестерпної спраги, потьмаренням свідомості, галюцинаціями, порушеннями ритму дихання. Підвищується температура тіла, розвивається "сольова гарячка ".
Зневоднення ендотеліальних клітин веде до збільшення проміжків між ними і, як наслідок, до збільшення проникності стінок судин. Це може бути причиною виходу з капілярів у тканини білків плазми крові та її формених елементів — розвиваються геморагії.
Захисно-компенсаторні реакції розвиваються припозаклітинній гіпергідрії:супроводжується збільшенням об'єму циркулюючої крові. Це веде до механічного розтягнення клітин передсердь, які у відповідь на це вивільняють у кров передсердний натрійуретичний гормон (атріопептин). Останній збільшує натрійурез і діурез, унаслідок чого зменшується об'єм циркулюючої крові. Збільшення об'єму циркулюючої крові є причиною зменшення імпульсації від волюморецепторів, у результаті чого зменшується секреція антидіуретичного гормону і зростає діурез.
133. Порушення обміну натрію і калію: причини, клінічні прояви та компенсаторно-пристосувальні механізми.
Натрій – основний катіон позаклітинної рідини.
Підвищення кількості натрію в плазмі крові (гіпернатріємія) спостерігають при олігурії або анурії будь-якого походження, гіперфункції кіркової речовини надниркових залоз (синдромі Кушінга, первинному альдостеронізмі), тривалому застосуванні кортикостероїдів, АКТГ, при парентеральних введеннях гіпертонічного розчину натрію, різкому обмеженні рідини.
Клініка:-сонливість;-судоми;-гіперрефлексія;-спрага,-нудота,-блювання;
Зниження концентрації натрію в плазмі крові (гіпонатріємія) обумовлено безсольовою дієтою, посиленим потовиділенням, тяжкими тривалими пологами, гострою і хронічною недостатністю надниркових залоз, діабетичним ацидозом, цирозом печінки, нефротичним синдромом, інтерстиціальним нефритом.
Клініка:-слабкість;-головокружіння,-втратаводи;-зниження тургору,сухість шкіри;-тахікардія;-артеріальна гіпотензія.
Калій є основним клітинним катіоном.
Підвищення кількості калію в плазмі крові (гіперкаліємія) спостерігається при гемолітичних анеміях, некрозі; порушенні виділення калію із сечею (олігурії, анурії, хронічному нефриті), зневодненні, анафілактичному шоці, парентеральному введенні розчинів, що містять іони К+, на тлі недостатності нирок;
Клініка:-загальна слабкість;-парестезії;-паралічі;-блювання;-біль у животі.
Зниження концентрації калію (гіпокаліємія) виникає при недостатньому вживанні калію з їжею, посиленому виділенні його із сечею, що може спостерігатися при гіперфункції кіркової речовини надниркових залоз і передньої частки гіпофіза, первинному та вторинному альдостеронізмі, посиленій секреції антидіуретичного гормону, діабетичному ацидозі, респіраторному алкалозі, парентеральному введенні великих кількостей рідини, що не містять калію, лікуванні антибіотиком гентаміцином, передозуванні АКТГ, препаратів кіркової речовини надниркових залоз, салуретиків.
Клініка:-гіпотонія;-брадикардія;-асистолія;-фібриляція шлуночків;-зміни об’єму серцевого викиду;-парастезії.
На ЕКГ знижується інтервал S — T, збільшується амплітуда зубця Р, зменшується (може збільшуватися) інтервал Р— Q, сплощується та інвертується зубець Т. За зниження концентрації К+ до 1,5 ммоль/л може виникати фібриляція шлуночків серця
134.Механізми регуляції водно-сольового обміну, компенсаторні реакції гіпо-та гіпергідрії.Гіперальдостеронізм,нецукровий діабет,гіпотиреоз.
Захисно-пристосувальні механізми при позаклітинному зневодненні:
1. перехід рідини з інтерстиціального сектора в судини,через зменшується гідростатичний тиск крові в капілярах, з одного боку, і збільшується онкотичний тиск крові внаслідок її згущення
2. Зменшення об'єму циркулюючої крові, пов'язане зі зневодненням, веде до збудження волюморецепторів і, в кінцевому підсумку, до збільшення секреції антидіуретичного гормону(збільшує реабсорбцію води в нирках, обмежуючи її втрату організмом)
3. Зменшення об'єму циркулюючої крові викликає активацію ренін-ангіотензинної системи і збільшення секреції альдостерону корою надниркових залоз. Це веде до збільшення реабсорбції іонів натрію в нирках і до нормалізації осмотичного тиску позаклітинної рідини.
4. У результаті зменшення артеріального тиску збуджуються барорецептори, що призводить до активації симпатоадреналової системи .
5. Зневоднення через центральні й периферичні механізми викликає відчуття спраги. У результаті формуються поведінкові реакції, спрямовані на пошук води й поповнення втраченої рідини.
Захисно-компенсаторні реакції розвиваються припозаклітинній гіпергідрії:супроводжується збільшенням об'єму циркулюючої крові. Це веде до механічного розтягнення клітин передсердь, які у відповідь на це вивільняють у кров передсердний натрійуретичний гормон (атріопептин). Останній збільшує натрійурез і діурез, унаслідок чого зменшується об'єм циркулюючої крові. Збільшення об'єму циркулюючої крові є причиною зменшення імпульсації від волюморецепторів, у результаті чого зменшується секреція антидіуретичного гормону і зростає діурез.
Гіперальдостеронізмом клінічний синдром з характерною підвищеною секрецією чи зміненим метаболізмом альдостерону і симптомами порушення водно-електролітного балансу та артеріальної гіпертонії.
1. Первинний альдостеронізм (аденома, аденоматоз, гіперплазія,
злоякісна аденома кіркової речовини).
2. Вторинний альдостеронізм (при повільному
розвитку серцевої недостатності, цирозі печінки, хронічному нефриті і
довготривалому застосуванні тіазидових діуретиків у хворих на гіпертонію та
пероральних контрацептивів, а також у вигляді пухлини Вільмса та
нефробластоми).Клініка:артеріальна гіпертензія постійна в межах 150-160 на 90-100
мм.рт.ст.,головний біль, зумовлений високим кров’яним тиском і
гіпергідратацією мозку носить інтенсивний характер.У міокарді внаслідок
дефіциту калію розвиваються дистрофічні зміни, що можуть бути причиною раптової
зупинки серця.
М’язова слабкісті, швидка втомлюваності
Ниркові прояви зумовлені втратами калію. При цьому
розвивається “калійпенічна нефропатія”, яка проявляється зниженням
концентраційної здатності нирок, поліурією, гіпо- і ізостенурією, ніктурією та
спрагою. Гіперкалійурія перешкоджає виведенню нирками Н+, а це
зумовлює лужну реакцію сечі.
Нецукровий діабет--внаслідок дефіциту антидіуретичного гормону,проявляється великою кількість сечі низької густини і компенсаторноюспрагою.Етіологія:пухлинигіпофіза ,гіпоталамуса,дія вірусів.Патогенез:Дефіцит АДГ--зниження реабсорбції води в канальцях--збільшення діурезу.У дітей проявляється затримкою фізичного і статевого росту
Гіпотиреоз - це дефіцит гормонів щитовидної залози. Симптоми у маленьких дітей включають порушення живлення і затримку фізичного розвитку. Ознаки у старших дітей і підлітків подібні таким у дорослих, але також включають низьке фізичний розвиток, пізніше статеве дозрівання абопоєднання.
Клініка:слабкість,брадикардія,набряки , закрепи,апатія.Вроджений гіпотиреоз – це хвороба. яка виникає у новонародженої дитини внаслідок недостатньої продукції гормонів, які виробляє щитоподібна з.тиреоїдних гормонів.У дітей затримка фізичного , статевого, розумового розвитку.Лікування замісна терапія
135.Набряки визначення, класифікація, причини , механізми розвитку.
Набряк – надмірне накопичення рідини у міжклітинному середовищі внаслідок порушення обміну води між кров’ю і тканинами – типовий патологічний процес. За етіологією: - набряк унаслідок ураження серця (застійний); - унаслідок ураження нирок; - унаслідок ураження печінки; - кахексичний (голодний); - запальний і токсичний; - нейрогенний; - алергічний.
Патогенезом:-мембраногенні;-лімфогенні;-мікседематозні;-онкотичні;-гідроосмотичні.
Фактори патогенезу набряків: - позитивний водний баланс (гіпергідрія – порушення функції нирок, вживання великої кількості осмотично активних речовин); - підвищення фільтраційного тиску (збільшення венозного тиску, розширення артеріол, звуження венул); - зниження градієнта осмотичного тиску між кров’ю і міжклітинною рідиною (гіпопротеїнемія, накопичення осмотично активних речовин); - підвищення проникності капілярів (вплив гуморальних факторів, порушення трофіки стінки); - порушення лімфовідтоку; - розлад нервової та гуморальної регуляції водно-мінерального обміну.
ТеоріяСтарлінга:вважав, щоперехідрідкоїчастиникровівміжтканиннийпростіріназадзумовленийтим, щовартеріальнійділянцікапіляраЕГТ (ефективнийгідростатичнийтиск) вищий, ніжЕОВС (ефективнаонкотичнавсмоктувальнасила), аувенознійділянці – навпаки. Тому розвиток набряку може бути зумовлений підвищенням гідростатичного тиску крові або зниженням її онкотичного тиску.
Патогенез серцевих набряків
(застійний набряк) – спричинюється здебільшого венозним застоєм і підвищенням венозного тиску, що супроводжується посиленням фільтрації плазми крові в капілярах. Гіпоксія, що розвивається, призводить до порушення трофіки і проникності стінки судин. Велике значення має також рефлекторно-ренін-адреналовий механізм затримання води: венозний застій – зменшення ниркового кровотоку – збільшення вироблення реніну – стимуляція секреції альдостерону, вторинний альдостеронізм – підвищення реабсорбції натрію та затримання води – гіпернатріємія – подразнення осморецепторів – посилення вироблення вазопресину – збільшення реабсорбції води.
Патогенез печінкових набряків
У розвитку печінкових набряків важливу роль відіграє гіпопротеїнемія,зумовлена порушенням синтезу білків у печінці.Певне значення має підвищення продукції або порушеня інактивації альдостерону.В розвитку асциту при цирозі печінки вирішальну роль відіграє утруднення кровообігу в печінці і підвищення гідростатичного тиску в системі ворітної вени.
Патогенез ниркових набряків.
Зменшення клубочкової фільтрації,що призводить до затримання води в орг-мі;Підвищується реабсорбція натрію в канальцях нефронів;Підвищення проникності стінок капілярів.Фактор гіпопротеїнемії (внаслідок протеїнурії),що поєднується із гіпер-волемією,що стимулює вироблення альдостерону.
Патогенез запальних набряків.
Порушення мікроциркуляції в осередку ураження і підвищення проникності стінки капілярів.У розвитку цих порушень відіграють роль звільнені вазо активніречовини-посередники:біогенніаміни(гістамін,серотонін),кініни(брадикінін),аденозин-фосфорнікислоти,похідніарахідонової кислоти(простагладини,лейкотрієни).
Патогенез алергічних набряків.
Алергічний набряк виникає у зв`язку із сенсибілізацією організму та алергічними реакціями (кропив`янка, алергіч. висип,ураження суглобів та ін.)Патогенез:порушенняпроникностістінкисудин,поруш.мікроциркуляції,утворення імунн. комплексів.
136. Механізми та прояви патологічноїдіїфакторіввиникненнянабряків (гідродинамічний, онкогенний, лімфогенний, мембраногенний). Гіпопротеїнемічнінабрякидитячоговіку.
Набряки — ценакопиченнярідини в тканинах організму і серознихпорожнинах.
Розрізняютьзагальні й місцевінабряки. Загальнінабряки є проявомпозаклітинної
Гіпергідрії, місцеві — пов'язані з порушенням балансу рідини в обмеженійділянцітканиниабо органа.
За етіологієювиділяютьнабряки:серцеві, ниркові, печінкові, кахектичні, запальні, алергічні, токсичні та ін.
Залежно від механізмів розвитку набряки можуть бути:
1) Гідростатичними;
2) Онкотичними;
3) Мікседематозними.
Гідростатичнінабрякивиникають у результатізбільшеннягідростатичноготиску в капілярах. Залежновід причин такого збільшенняможнавиділити:
А) гіперволемічні;
Б) застійні;
В) мікроциркуляторнінабряки.
Онкотичнінабрякивиникаютьунаслідокзмінонкотичноготиску в капілярахабоінтерстиціальнійрідині. У цюгрупувходять:
А) гіпопротеїнемічні;
Б) мембраногенні;
В) лімфогеннінабряки.
Мембраногеннінабрякивиникаютьунаслідокпідвищенняпроникностісстіноксудин (див. Розд. 14). Збільшенняпроникностісудин є причиною виходубілківплазмикрові в міжклітиннийпростір, унаслідокцьогозростаєтканиннийонкотичнийтиск і вода переходить ізкровоноснихсудин в інтерстицій.Зазначениймеханізм є провідним у розвиткуалергічних, запальних, токсичнихнабряків.
Лімфогеннінабрякивиникаютьунаслідокпорушеньлімфоутворення і лімфовідтоку. При цьомупорушуєтьсявиведення з лімфоюбілків, що в норміфільтруються у тканину, і, як наслідок, збільшуєтьсятканиннийонкотичнийтиск.Серед причин розвиткулімфогеннихнабряківвартовиділитиздавленнялімфатичнихсудинрубцем; збільшення центрального венозного тиску
137. Поняття про неімунну водянку плода, їїетіопатогенез.
Неімуннаводянка плода, розвитокякої не пов’язаний з гемолітичною хворобою.Вона є дужесерйознимвнутрішньоутробнимзахворюванням, котрехарактеризуєтьсявираженоюзагальноюгідратацією плоду внаслідокнадлишковогопозаклітинногонакопиченнярідини в тканинах і серознихппорожнинах при відсутностіознакімунноїссенсибілізції. При народженнідитини з даноюпатологієюзвертають на себе увагувираженітотальнінабрякивнаслідокнакопиченнярідини у всіхпорожнинахтіла. Найбільшу ж кількістьрідинивиявляють у плевральній та черевнійпорожнинах. Досить часто за таких умов життядитиниврятувати не вдається.
Патогенез водянки:
Поява водянки
обумовлена причинами, які призводять до змін в розподілі рідини між
внутрішньо-судинними та інтерстиціальними позаклітинними просторами. У
більшості випадків набряк виникає при зниженні білкового складу сироватки, що
призводить до зниження тиску у капілярах.
Набряки
можуть утворюватись при змінах у вмісті електролітів в поза-судинній і судинній
рідині, що створює різницю в остмотичному тиску.
У
недоношених новонароджених набряк спричиняється зниженням здатності виведення
води та натрію, але в деяких випадках походження його невідоме.
У
новонароджених набряк голови і шиї може бути наслідком обвиття шиї пуповиною, а
транзиторні обмежені набряки кистей та ступнів виникають внаслідок
внутрішньоутробного стиснення. Неімунна водянка може супроводжувати серцеву
недостатність, пов'язану з вродженими захворюваннями серця, навіть при
відсутності шумів.
Майже
в 44% причину виникнення неімунної водянки встановити не вдається і її вважають
ідіопатичним станом.
138. Поняття про синдром імунної водянки плода, йогоетіопатогенез.
Імунна водянка плода—форма водянки плода, супроводжуючинадлишковимскопченнямплодноїрідини в не судиннихвідділах і порожнинахтіла, через несумісністькровіматері і плода по резус фактору
139. Поняття про «невідчутну» втратурідини у новонародженихнемовлят: етіологічніфактори, механізми, принципипрофілактики.
Втрати води з організмуновонародженогозалежатьвідступеняйогозрілості, умов навколишньогосередовища, наявностіта характеру захворювання і здійснюються через перспірацію злегень і шкіру («невідчутнівтрати»), з калом і сечею.
Обов’язковослідвраховувативтратуорганізмом води, обумовленувипаровуванням з поверхнітіла і дихальнихшляхівновонародженого, або так звані «невідчутнівтрати», якізначновідрізняютьсявіданалогічних величин дорослоїлюдини.встановлено, щовтрати води через легені і шкіру у зздоровоїдоношеноїдитинисягають 20-35 мл/кг на добу (в середньому 30 мл/кг/добу). Відмінноюрисою водного обмінуновонародженихє те, що у них дужевисокевиведення води через легені і шкіру (в порівняні з дорослоюлюдиною). Величина цихвтратоберненопропорційнамасітіла при народженні та гістаційномувіку, при цьомуприблизно 30% невідчутнихвтрат у новонародженихвідбувається з дихальнихшляхів і 70% - шляхом випаровуваннязішкіри, тобтоневідчутнівтрати з диханнямудвічінижчі, ніжчерез шкіру. Хоча у новонародженихкількістьпотовихзалоз в 6 разівбільшаніж у дорослих, максимальна потовареакція у відповідь на тепловустимуляцію становить лише 1/3 віддорослого. Випаровування води зішкіри особливо значне в недоношенихдіітей. Так об'ємневідчутнихвтрат у новонароджених з масоютілапонад 1500 г становить 40-50 мл/кг/добу, а у дітей з масоюТіла 1500 г і меншеколиваєтьсявід 55 до 90 мл/кг на добу. встановлено, що при дихальнійнедостатності ІІ-ІІІ ступеняперспіраційнівтратирідинизбільшуються в середньому на 10-20 мл/кг/добу, а при частотідиханнябільше 60 за хвилину накожні 10 вище 60 зростають на 10 мл/ кг/ добу. Підвищеннятемпературитілавище 37,5°С протягомбільше 6 годин потребуєдобавлення до необхідногооб’ємурідинище 10 мл/кг/добу
Зменшитиневідчутнівтратиможна шляхом зволоження і зігрівання до термонейтральногорівняповітряно-кисневусуміш, яка надходить в кювез, або в аппарат для ШВЛ. Крім того, обмежуютьвипаровуваннязішкіри шапочка на головідитини та Створенняекрану над тулубом і кінцівками за типом «теплиці».
140. Причини та патогенез гострого запального набрякулегеньдитячоговіку (респіраторногодистрес-синдрому).
Респіраторнийдистрес синдром (РДС), або хвороба гіалінових мембран, – гострепорушенняфункціїдихання, щорозвиваєтьсяпереважно у недоношенихдітей у першігодинижиття, обумовлененезрілістюлегень, дефіцитомсурфактанту.
Частота захворюваннязалежитьвідгестаційноговікуновонародженого. При гестаційномувіціменше 26 тижнів вона складає 90%; 28-29 тижнів –
50-70%; 30-32 тижні – 20%; у доношенихдітей – менше 1%.
Етіологія, патогенез
Причина захворювання – дефіцитсурфактанту, якийможе бути обумовлений: недостатнім синтезом сурфактанту; порушеннямвикидусурфактанту; підвищенимруйнуваннямсурфактанту; інгібуваннямсурфактанту.
Основніфакториризикурозвитку РДС:
- недоношеність та незрілістьновонародженого;
- внутрішньоутробнагіпоксія плоду;
- асфіксіяновонародженого;
- внутрішньоутробнеінфікування;
- кесарськийрозтин;
- цукровийдіабет у матері, якийвикликає ацидоз плоду;
- багатопліднавагітність;
- переохолодженняновонародженого;
- гіповолемія плоду та новонародженого.
Сурфактантлегень – цеповерхнево-активна речовина, яка вкриваєповерхність альвеол, та синтезуєтьсяальвеоцитами ІІ типу й клітинами Кларка; складається з ліпідів (90%) та неплазменнихсурфактант-асоційованихпротеїнів (10%). Продукціялегеневогосурфактантупочинається на
20-24 тижнівнутрішньоутробногорозвитку, до 34-35 тижня шлях синтезу йогодуженестійкий і легко порушується при гіпоксії, гіпотермії та ацидозі
141. Ацидоз: визначення поняття, класифікація, причини розвитку. Компенсаторні та патологічні реакції. Показники кислотно-основного стану при різних видах ацидозу, принципи корекції.
Ацидоз – це зміщення кислотно-основної рівноваги (КОР) в бік кислотності (pH<7).
Класифікація:
1. В залежності від впливу на КОР: компенсовані (коли рН артеріальної крові не виходить за межі норми, 7,37 – 7,43) або декомпенсованими (коли буферні системи не спроможні підтримувати рН).
2. В залежності від причини змін КОР: газові (обумовлені збільшенням вмісту Н2СО3) та негазові (через зменшенням вмісту бікарбонату НСО3-).
Причини:
1. Газового: ендогенний (порушення виведення бікарбонату з організму при всіх видах гіповентеляції – недостатність зов дих) та екзогенний (вдихання великої к-сті бікарбонату)
2. Негазовий: метаболічний (накопичення кислот-метаболітів при ЦД, гіпоксії, голодуванні, ХНН, подагрі, тощо), екзогенний (надходження в організм кислих нелетких кислот при тривалому вжив кислих продуктів, прийм ліків або отруєні) та видільний (втрачання бікарбонату через порушення реабсорбції в нирках та ШКТ).
Компенсаторні реакції
1. Газового: спрямовані на зб концентрації бікарбонату (ацидогенез), забезпечується нирками шляхом підкислення сечі та реабс НСО3-
2. Негазового: дихальні зміни (зб концен Н+ - збудж дих центру – гіпервентиляція – виведення СО2 з організму) та ниркові ( зменш рН – акт ацидогенезу в канальцях нирок).
Патологічні реакції: однакові при всіх ацидозах, обумовлені підв конц СО2 (збудження дихального центра та задишка, розширення мозкових судин та звуж судин м'язів та нирок, зміщення кривої дисоціації гемоглобіну вправо) та безпосередньо ацидозом (пригн скор функц міокарду через блокаду Са-каналів воднем, порушення конформації білків, зб прон мембран, акт ліз ферментів, ацидотична кома).
Кореляція:
1. Газового: відновлення напруги вуглекислого газу в крові шляхом його виведення з організму – відновлюють зов дихання або виводять з приміщення багатого на вуглекислий газ.
2. Негазового: нормалізація концентрації бікарбонату в плазмі крові – в/в NaHCO3
SB – рівень бікарбонату


142. Негазовий ацидоз (метаболічний, видільний, екзогенний). Форми канальцевого ацидозу. Синдром Баттлера-Олбрайта.
Причини негазового ацидозу:
1. Метаболічний (накопичення кислот-метаболітів при ЦД, гіпоксії, голодуванні, ХНН, подагрі, тощо)
2. Екзогенний (надходження в організм кислих нелетких кислот при тривалому вжив кислих продуктів, прийм ліків або отруєні)
3. Видільний (втрачання бікарбонату через порушення реабсорбції в нирках та ШКТ)
a. Проксимальний/ ацидоз 2 типу (перв поруш реабсорбції бікарбонату в проксим звивистих канальцях)
b. Дистальний/ацидоз 1 типу/синдром Баттлера-Олбрайта (перв поруш ацидогенезу в дистальних звивистих канальців – організм не витримує навант кислотами-метаболітами
c. Видільний (внаслідок порушення реабсорбції бікарбонату з підшлунк та кишков соком при діареї, при гіперсальвації, бо слина багата бікарбонітами)
143.Алкалоз: визначення поняття, класифікація, причини розвитку. Компенсаторні та патологічні реакції. Показники кислотно-основного стану при різних видах алкалозу, принципи корекції.
Алкалоз – це зміщення КОР в бік лужності.
Класифікація:
1. В залежності від впливу на КОР: компенсовані (коли рН артеріальної крові не виходить за межі норми, 7,37 – 7,43) або декомпенсованими (коли буферні системи не спроможні підтримувати рН).
2. В залежності від причини змін КОР: газові (обумовлені зменшенням вмісту Н2СО3) та негазові (через збільшенням вмісту бікарбонату НСО3-).
Причини:
1. Газовий: гіпервентиляція легень, що обумовлена безпосеред. зб дих центру при пухлині/енцефаліті, рефлект зб дих центру при гірській хворобі/гарячці та неправильному штучному диханні
2. Негазовий: гіпохлоремічний (втрата Сl при блюванні – вона не потрапляє ДПК – не стим утв секретину – пригнічення секрец панкреат соку – бікарбонат затримується в організмі – алкалоз), гіпокаліємічний (зниження конц К+ через гіперальдесторинізм –зменшення К в клітині – поруш обміну К на Н+ - накопичення водню в клітині – алкалоз), гіпернатріємічний ( надходження екзогенних основ).
Компенсаторні реакції:
1. Газовий: зниження вмісту бікарбонату, забезпечується нирками (змен рСО2 – зм інтенсивність ацидогенезу в проксим канал нефронів – зм реабсорбції бікарбонату – зб рН сечі та зм конц бікарбонату в плазмі крові-зм рН крові до норми)
2. Негазовий: зб напруги вуглекислого газу, забезпечується зов дих ( пригніч дих центру – пригн вентиляції – накопичується СО2)
Пат зміни: обумовлені гіпокапнією ( розширення периферичних судин – гіпотензія, звуж мозкових та коронар судин – гіпоксія мозку та серця, зміщ кривої дисоціації оксигемоглобіну вліво – не віддає кисень) та власне алкалозом (зміна конформації білків, зб їх гідрофільності – набряк, порушення окисного фосфорилювання через поруш градієнту Н+)
Кореляція:
1. Газового: боротьба з гіпокапнією та відновлення напруги вугл газу в крові – викор газових сумішей (напр. карбоген)
2. Не газового: відновлення концентрації іонів ( розчин NH4Cl, KCl, інгібітори карбонгідрази для приг ацидогенезу та реабс бікарбонату)


SB – рівень бікарбонату
144. Особливості підтримання кислотно-основного стану у дітей.
У дітей раннього віку спостерігається виражена схильність до ацидозу (особливо метаболічного – вродженийлактато-ацидоз, щообумовлений дефектом ключових ферментів), особливо у новонароджених. Це можна пояснити інтенсивністю у них біоенергетичних процесів, обмеженими функціональними можливостями нирок і інших органів і систем, які беруть участь в регуляції кислотно-лужної рівноваги.
ДРУГИЙ СЕМЕСТР
1. Анемії: визначення поняття, класифікація анемій (за етіологією, патогенезом, типом еритропоезу, регенераторною здатністю кісткового мозку, колірним показником, змінами розмірів еритроцитів). Навести приклади гіпо- та гіперхромних анемій.
Анемії – це клінічно-гематологічний синдромабо самостійна хвороба, що характеризується зменшенням кількості еритроцитів та/або зменшення гемоглобіну, а також якісними змінами еритроцитів.
Класифікації:
1. Патогенетична класифікація: постгеморагічні (після кровотечі), гемолітичні (після значного лізису еритроцитів) та обум поруш еритропоезу
2. За етіологією: спадкові (таласемії) або набуті (хронічна постгеморагічна)
3. За інтенс регенерації: регенераторні (гостра постгеморагічна), гіперррегенераторна (набута гемолітична), гіпорегенераторна (залізодеф.) та арегенераторна (апластична)
4. За колірним показником: нормохромна (КП=0,85-1, гостра постгеморагічна), гіпохромна (КП<0,85, залізодефіцитна) та гіперхромна (КП>1, мегалобластична анемія)
5. За типом кровотворення: з еритробластним типом поезу та з мегалобластним
6. За розміром еритроцитів:мікроцитарними, нормоцитарними та макроцитарними
7. За клін перебігом: гостра та хронічна
2.Дегенеративні та регенеративні форми еритроцитів. Етіологія, патогенез, гематологічна характеристика постгеморагічної анемії (гострої і хронічної)
До регенеративних форм еритроцитів належать ретикулоцити, які виявляються при суправітально забарвленні мазка крові (в нормі їх число становить 0,5-2%), поліхроматофільние еритроцити (еквіваленти ретикулоцитів, які виявляються при фарбуванні мазка за Романовським), ацидофільні та поліхроматофільние нормобласти (в нормі їх в крові немає, вони знаходяться в кістковому мозку). Клітини патологічної регенерації еритроцитів - це мегалоціти і ацидофільні, поліхроматофільние, базофільні мегалобластов.
Дегенеративні зміни еритроцитів можуть полягати у зміні:
1. величини еритроцитів (анізоцитоз), що проявляється в наявності макроцитів і мікроцітов;
2. форми еритроцитів (пойкилоцитоз), коли спостерігаються в мазку крові еритроцити грушоподібної форми, витягнуті в довжину, серповидні, овальні, сферичні;
анізохромія;
3. забарвлення еритроцитів, в залежності від вмісту в них гемоглобіну - переважання інтенсивно забарвлених гіперхромних еритроцитів, бліднокрашених гіпохромних еритроцитів, анулоцітов поряд з нормохромного еритроцитами, що при вираженому відмінності в забарвленні прийнято називати анізохромія;
4. наявності патологічних включень в еритроцитах - тілець Жолли, кілець Кебота, базофільною зернистості, і ряд інших.
І Постгеморагічні анемії - розвіваються внаслідок гострої чи хронічної крововтраті. Гостра - Регенераторна анемія. Хрон - гіпоренегераторна. ІІ Гемолітічні - что вінікають внаслідок підвіщеного ерітродіурезу, коли гемоліз переважає Утворення. Набутів - Регенераторна, Спадкового - гіперрегенераторна. ІІІ Дісерітропоетічні – гіпорегенераторні.
3.Гемолітичні анемії. Класифікація гемолітичних анемій за етіологією, за місцем гемолізу. Механізми гемолізу
гостра: після швидкої масивної крововтрати внасл поранення судин чи руйнування стінки судин пат процесом;
Гостра крововтрата. 1-ші години. Компенсаторні р-ції: рефлект спазм судин (підвищення опору в судинах внутрішніх органів і шкіри) , вихід крові з депо в кровоносне русло, внаслідок чого підвищується артеріальний тиск, до певної міри відновлюються об'єм циркулюючої крові і кровопостачання життєво важливих органів; рефлекторне почастішання і посилення серцевих скорочень; вихід міжтканинної рідини в судини; рефлекторне почастішання і поглиблення дихання, сприяюче усуненню дефіциту кисню в організмі; підвищення здатності гемоглобіну віддавати кисень тканинам; підвищення згортання крові, що припиняє кровотечу.
К-ть еритроцитів, Hb в одиниці об‘єму – N, нормоцитемічна гіповолемія, зменш абс маси еритроц. Еритроц без якісних змін.
За 2-3 дні після спинення: еритр і Hb в од об‘єму зменш за рахунок надходження тканинної р-ни в судини, КП – N.
4-5 дні: збільш проліфер кл еритроцитарного ростка км під впливом еритропоетину (внасл гіпоксії). В крові зрост поліхром еритр, ретикулоц, є поодинокі нормобласти ( регенераторна анемія). КП – гіпохромна.
Патогенез:
Початкова стадія характеризується зменшенням об'єму циркулюючої крові - простий гиповолемией, зниженням серцевого викиду, падінням артеріального тиску, гіпоксією переважно циркуляторного типу.
• Компенсаторна стадія обумовлена включенням потужного комплексу захисно-пристосувальних реакцій, спрямованих на відновлення об'єму крові, нормалізацію гемодинаміки, кисневого забезпечення організму.
Термінові Механізми компенсації:
1. рефлекторний спазм кровоносних судин (за винятком головного мозку і серця), вихід крові з депо;
2. рефлекторне почастішання і посилення серцевих скорочень;
3. надходження міжтканинної рідини в судини;
4. рефлекторне почастішання і поглиблення дихання;
5. підвищення здатності гемоглобіну віддавати кисень тканинам;
4.Набуті гемолітичні анемії. Класифікація за етіологією, механізми гемолізу.
хронічна: внасл повторних крововтрат, зумовленіх Ураження кров Судін за питань комерційної торгівлі хвороб чи порушеннях Тромбоцит-судинного и коагуляції гемостазу (геморагічній синдром).
Зменшіть запасів Fe в про-мі при повторних крововтрата (вінікає залізодефіцітна), низька р-нь Hb, знижено КП, з'явилися в мвзку крови гіпсохромніх Еритреї, мікроцітів. При прігніченні кровотворення - гіпорегенераторна.
Гемолітичні – що виникають внаслідок підвищеного еритродіурезу, коли гемоліз переважає утворення.
Класифікація.
І Набута: 1) токсична – дія інф ф-рів, хім. Реч-н різної природи; 2) імунна (ізо-, гетеро-, авто-) – під впливом комплексу АГ-антиеритроцитарне АТ; 3) механічна – мех. Ушкодження еритр; 4) набута мембранопатія – пов‘язана з сомат мутацією, коли в км утв клон еритр з дефектом стр-ри мембрани.
ІІ Спадкова: 1) спадкова мембранопатія, 2) ферментопатія, 3) гемоглобінопатія (а) зумовлена ген дефектом перв стр-ри ланцюгів глобіну, б) – зум порушенням синтезу а- чи в- ланцюгів глобіну)
Набута. Еритробластна, регенераторна, нормо- чи гіпохромна, псевдогіперхромна (абсорбція Hb на еритр). У мазку: високий ретикулоцитоз, пойкілоцитоз, анізоцитоз. При геполіт хв. ново нар – еритробласти, нормобласти.
Спадкова. Гіперрегенераторна, з неефективним еритропоезом (в км руйн ядерні форми). Мазок: ретикулоцитоз, поліхроматофілія, поодинокі нормобласти; дегенеративно змінені ф-ми (хв. Мінковського-Шоффара – мікросфероцити, Hb S-гемоглобінопатії – дрепаноцити (серпоподібні), таласемія – мішене подібні, еритр з базофільною зернистістю). Може гіпо- чи арегенераторна.
Гемолітична хвороба – хвороба, що виникає в результаті гемолізу еритр плода і новонародженого, що викликаний АТ матері.
Варіанти – резус-конфлікт, АВ0 конфлікт.
Резус-конфлікт. При вагітності Rh- матері Rh+ плодом. Спочатку – імунізація матері Rh+ еритр плода. У відповідь на їх поступлення в о-мі матері синтезуються АТ проти D-АГ. Ці АТ (IgG) проникають через плаценту, викликають гемоліз еритр.
АВ0-конфлікт. Частіше: мати – 0(І), плід – А(ІІ) чи В(ІІІ). Норм ізоаглютиніни в с-мі АВ0 належать до класу IgМ. Ці АТ не прон через плаценту. У 10% людей з 0(І) є АТ проти аглютиногенів А і В, що представлені IgG. Їх наявність не залежить від попередньої імунізації. Ці аглютиніни прон через плаценту, викл гемоліз еритр плода з А(ІІ), В(ІІІ).
Гемоліз відбувається не лише усередині судин, а і в печінці, селезінці.
5. Спадкові гемолітичні анемії Спадкові мемранопатії. Причини, механізми, клінічні прояви
Мембранопатії. Основу цієї групи анемій становлять дефекти мембран еритроцитів.
Спадкові мембранопатії можуть бути обумовлені двома групами дефектів ери-троцитарної мембрани. З урахуванням цього виділяють дві групи мембранопатій.
1. Мембранопатії, обумовлені порушеннями мембранних білків. До них, зокрема, відносять:
а) мікросфероцитарну анемію Мінковського-Шоффара;
б) овалоклітинну гемолітичну анемію (еліптоцитоз, овалоцитоз);
в) стоматоцитоз.
2. Мембранопатії, пов'язані з порушеннями мембранних ліпідів. У цю групу входять:
а) акантоцитоз (є проявом абеталіпопротеїнемії);
б) анемія, обумовлена дефіцитом ЛХ4Г(лецитин-холестерол-ацилтрансферази).
Анемія Мінковського-Шоффара є спадковою, ендоеритроцитарною (мембранопатія) гемолітичною анемією з внутрішньоклітинним гемолізом. Тип спадкування - аутосомно-домінантний. Спадковий дефект виникає в мембранному білку еритроцитів - спектріні. Унаслідок цього значно збільшується проникність еритроцитарної мембрани для іонів натрію. Натрій і вода переходять з плазми всередину еритроцитів. Це врікликає активацію Na-K-насосів і гліколізу, завдяки чому еритроцити підтримують у цих умовах свій об'єм. Якщо ж зазначені механізми виявляються функціонально недостатніми, то об'єм еритроцитів збільшується й вони набувають сферичної форми, тобто перетворюються на сфероціїти (кульки). Сфсроцити втрачають свою пластичність, а отже, і здатність до деформації. Через це вони не можуть проходити через вузькі міжендотеліальні щілини венозних синусів селезінки й на тривалий час затримуються в ній. Макрофаги селезінки "відкушують" частину мембрани еритроцитів і перетворюють останніх на мікросфероцити. При наступних проходженнях мікросфероцитів через селезінку макрофаги повністю фагоцитують змінені еритроцити -відбувається внутрішньоклітинний гемоліз. Таким чином, тривалість життя еритроцитів зменшується до 8-12 діб замість 120. З урахуванням патогенезу зазначеної анемії непоганий лікувальний ефект має видалення селезінки — спленектомія Клініка. Хвороба вперше проявляється у дитячому або, найчастіше, підлітковому віці різного ступеня анемією, жовтяницею, збільшенням селезінки та схильністю до утворення каменів жовчного міхура. Ступінь вираження клінічних проявів ССц має індивідуальний характер і варіює залежно від характеру дефекту білків мембрани і, отже, тяжкості перебігу хвороби. Залежно від тяжкості перебігу гемолізу виділено чотири форми ССц (прихована (латентна), легка типова (середньої тяжкості), тяжка).
Спадковий еліптоцитоз (синонім - овалоцитоз) - спадково детерміноване захворювання, що передається по аутосомно-домінантним типом успадкування, пов’язане зі зменшенням вмісту АТФ та 2-3-ДФГ у мембрані еритроцитів. Розвиваючись у своїй аномалія еритроцитів характеризується розладом структури мембрани еритроцитів. Клініка. У більшості випадків дана аномалія не дає ніяких клінічних проявів, однак у частини хворих хвороба супроводжується гемолітичною анемією з внутрішньоклітинним розпадом еритроцитів, переважно в селезінці. У нормі вміст овальних еритроцитів у здорових людей може досягати 10%. У хворих спадковим еліптоцитозом еліптоцити становлять 25-75%. Частота гена еліптоцитоза в людській популяції становить 0,02-0,04%, захворювання зустрічається з тією ж частотою поширеності, як і мікросфероцитоз, однак діагностується рідше, оскільки у значної частини носіїв відсутні будь-які прояви захворювання. В загальному аналізі кров – анемія, ретикулоцитоз, збільшення концентрації білірубіну за рахунок непрямої фракції. Еритроцити еліптоїдної форми, анізо- та пойкілоцитоз. Осмотична резистентність еритроцитів значно знижена. У кістковому мозку спостерігається подразнення еритроїдного паростка. Захворювання має спокійний, хронічний перебіг і лише в окремих випадках з невідомих причин набуває характеру гемолітичних криз із внутрішньо судинним гемолізом. Розвивається анемія, жовтяниця, спленектомія.
6. Спадкові гемолітичні анемії. Ферментопатії. Причини, механізми, клінічні приклади
Ферментопатії (ензимопатії). Анемії цієї групи обумовлені спадковими порушеннями ферментів еритроцитів.
Причиною розвитку спадкових ферментопатій можуть бути порушення таких ферментних систем еритроцитів.
1. Дефіцит ферментів пентозного циклу. Найпоширенішою ферментопатією є глюкозо-6-фосфатдегідрогеназодефіцитна анемія, обумовлена повною відсутністю або значним зменшенням активності глюкозо-6-фосфатдегідрогенази. Відомо близько 250 мутантних форм цього ферменту.
2. Дефіцит ферментів гліколізу. Описано спадкові дефекти 11 з 13 ферментів гліколізу. Найбільш поширеним є дефіцит піруеаткінази.
Основою патогенезу анемій, що виникають у результаті порушень реакцій гліколізу, є дефіцит АТФ, що закономірно призводить до порушення енергозабезпечення Na-K-насосів плазматичних мембран. Наслідком цього є збільшення концентрації іонів натрію і, як результат, осмотичного тиску всередині еритроцитів, що, у свою чергу, викликає надходження води в клітини. При цьому еритроцити набухають і перетворюються на сфероцити, які зазнають фагоцитозу з боку макрофагів.
3. Дефіцит ферментів циклу глютатіону (глютатіонсинтетази, глютатіонредукта-зи, глютатіонпероксидази).
Порушення циклу глютатіону призводять до пригнічення роботи глютатіонпе-роксидазної антиоксидантної системи, наслідком чого є активація процесів пе-роксидного окиснення ліпідів в еритроцитах. Реакції вільнорадикального окис-нення ліпідів мембран порушують їх бар'єрні властивості й значно збільшують проникність еритроцитарної мембрани до іонів, зокрема, іонів натрію. Останні за градієнтом концентрації надходять у клітини, збільшуючи осмотичний тиск усередині еритроцитів. Подальші процеси аналогічні тим, які розвиваються при інших описаних тут ферментопатіях.
4. Дефіцит ферментів утилізації АТФ. Прикладом цієї групи ферментопатій є дефіцит білкових компонентів Na-K-АТФ-ази еритроцитарної мембрани. Первинні порушення роботи Na-K-насосів еритроцитів призводять до збільшення концентрації іонів натрію в клітинах з усіма наслідками, що звідси випливають.
Глюкозо-6-фосфатдегідрогеназодефіцитна анемія є спадково обумовленою гемолітичною анемією, ендоеритроцитарною (ферментопатія), з внутрішньосудинним гемолізом. Тип спадкування - зчеплений з Х-хромосомою.
У патогенезі цієї анемії центральне місце посідають такі процеси. Дефіцит глюкозо-6-фосфатдегідрогенази є причиною порушень реакцій пентозного циклу, внаслідок чого зменшується утворення НАДФН і відновлення (регенерація) глютатіону в еритроцитах. Це, у свою чергу, призводить до зниження активності антиоксидантної глютатіонпероксидазної системи і збільшення небезпеки активації вільнорадикального окиснення. Цим, зокрема, пояснюється збільшення чутливості еритроцитів до дії екзогенних окислювачів (лікарських препаратів, токсичних продуктів рослинного походження) і активація пероксидного окиснення ліпідів мембран. Останнє призводить до збільшення проникності еритроцитарних мембран і надходження в клітини іонів натрію й води. Розвивається набухання й набряк еритроцитів, а потім - їх внутрішньо судинний гемоліз.
*Під дією пероксидів виникає окисна денатурація гемоглобіну з утворенням тілець Гейнца-Ерліха.
Однією з клінічних форм глюкозо-6-фосфатдегідрогеназодефіцитної анемії є фавізм, що виникає при вживанні бобів Vicia fava, які містять токсичні продукти з вираженими окисними властивостями.
7. Спадкові гемолітичні анемії. Гемоглобінопатії. Серпоподібно клітинні анемії ( генетичний дефект, що її спричинює, тип спадкування, патогенез, картина крові)
Спадкова: 1) спадкова мембранопатія; 2) ферментопатія; 3) гемоглобінопатія: а) зумовлена ген дефектом первстр-ри ланцюгів глобіну, б) зум порушенням синтезу а- чи в- ланцюгів глобіну.
Спадкова. Гіперрегенераторна, з неефективним еритропоезом (в км руйн ядерні форми). Мазок: ретикулоцитоз, поліхроматофілія, поодинокі нормобласти; дегенеративно змінені ф-ми (хв. Мінковського-Шоффара – мікросфероцити, Hb S-гемоглобінопатії – дрепаноцити (серпоподібні), таласемія – мішене подібні, еритр з базофільною зернистістю). Може гіпо- чи арегенераторна.
Гемоглобінопатія — спадкова або вроджена зміна структури білка гемоглобіну, зазвичай приводить до змін в його функції транспорту кисню або у будові та функції еритроцитів.
8. Спадкові гемолітичні анемії. Патологічні форми еритроцитів, характерні для різних видів гемолітичних анемії. Загальні прояви та механізми компенсації.
До регенеративних форм еритроцитів належать ретикулоцити, які виявляються при суправітально забарвленні мазка крові (в нормі їх число становить 0,5-2%), поліхроматофільние еритроцити (еквіваленти ретикулоцитів, які виявляються при фарбуванні мазка за Романовським), ацидофільні та поліхроматофільниенормобласти (в нормі їх в крові немає, вони знаходяться в кістковому мозку).
Клітини патологічної регенерації еритроцитів - це мегалоціти і ацидофільні, поліхроматофільние, базофільнімегалобластов. Дегенеративні зміни еритроцитів можуть полягати у зміні:
1. величини еритроцитів (анізоцитоз), що проявляється в наявності макроцитів і мікроцітов;
2. форми еритроцитів (пойкилоцитоз), коли спостерігаються в мазку крові еритроцити грушоподібної форми, витягнуті в довжину, серповидні, овальні, сферичні;
3. забарвлення еритроцитів, в залежності від вмісту в них гемоглобіну - переважання інтенсивно забарвлених гіперхромних еритроцитів, бліднокрашенихгіпохромних еритроцитів, анулоцітов поряд з нормохромного еритроцитами, що при вираженому відмінності в забарвленні прийнято називати анізохромія;
4. наявності патологічних включень в еритроцитах - тілець Жолли, кілецьКебота, базофільною зернистості, і ряд інших.
Гемолітічні - чтовінікають внаслідок підвіщеногоерітродіурезу, коли гемоліз переважає утворення.Набута - Регенераторна, Спадкового - гіперрегенераторна.
9. Патогенез та прояв гемолітичної хвороби плода\ новонародженого (резус – конфлікт). Профілактика резус- конфлікту.
Гемолітична
хвороба – хвороба, що виникає в результаті гемолізу еритр плода і
новонародженого, що викликаний АТ матері.
Варіанти – резус-конфлікт, АВ0 конфлікт.
Резус-конфлікт. При вагітності Rh- матері Rh+ плодом.
Спочатку – імунізація матері Rh+ еритр плода. У відповідь на їх поступлення в
о-мі матері синтезуються АТ проти D-АГ. Ці АТ (IgG) проникають через плаценту,
викликають гемоліз еритр.
АВ0-конфлікт. Частіше: мати – 0(І), плід – А(ІІ)
чи В(ІІІ). Норм ізоаглютиніни в с-мі АВ0 належать до класу IgМ. Ці АТ не прон
через плаценту. У 10% людей з 0(І) є АТ проти аглютиногенів А і В, що
представлені IgG. Їх наявність не залежить від попередньої імунізації. Ці
аглютиніни прон через плаценту, викл гемоліз еритр плода з А(ІІ), В(ІІІ).
Гемоліз відбувається не лише усередині судин, а
і в печінці, селезінці.
Профілактика. Планова допологова профілактика проводиться всім резус-негативним жінкам на 28-30 тижні вагітності (одна внутрішньом'язова ін'єкція анти-D-імуноглобуліну).
Екстрена допологова профілактика проводиться в разі виникнення потенційно небезпечних ситуацій, які багаторазово підвищують ризик розвитку резус-конфлікту.
10. Класифікації анемій, пов’язаних з порушеннями еритропоезу ( дефіцитні, дисрегуляторні, гіпо-, апластичні та ін.), загальна характеристика причин і механізмів розвитку
Є набуті, спадкові. За мех. Порушення еритропоезу: 1) дисрегуляторна – внаслідок зниження синтезу еритропоетину, підвищ синтезу його інгібіторів (хрон захв нирок, гіпоф-ція гіпофізу, ЩЗ); 2) дефіцитна – нестача р-н, потрібних для утв еритроцитів (залізо, В12-, фолієво-, білководефіцитні); 3) ферментопатії (поруш у синтезі порфірину і гему); 4) гіпо(а)пластична – ураження еритр ростка КМ на тлі різкого пригнічення КМ кровотворення; 5) мета пластична (мієлофтизична) – після заміщення чи витіснення еритр ростка іншою тканиною.
Тип еритропоезу: еритробластні, мегалобластні. Клініка: переважно хронічні
Апластична (АА) – анемія, внасл якої уражується еритр росток КМ на тлі глибокого пригнічення кровотворення, різко знижується продукція еритроцитів, гранулоцитів, тромбоцитів (панцитопенія). Належить до синдрому недостатності КМ.
Патогенез. Мех. Пригнічення гемо(еритро)поезу: 1) зменш к-ті кл-попередників еритр ряду під прямою ушкодж дією етіологічних ф-рів на СК КМ чи їх мутації – зменш мітотична активність кровотворних кл, поруш синтез Hb (збільш вмісту HbF при анемії Фанконі); 2) імунне ушкодж АТ і Т-лімфоцитами еритропоетинчутливихкл чи кл-попередників мієлоплезу в КМ, еритропоетину і його рецепторів; 3) неповноцінність стромальногомікрооточенняеритропоетичнихкл, - поруш їх проліф ф-ція і здатність до диференціювання.
КМ: різке збіднення на клітинні елементи, кровотворна тк заміщується жировою. Неефективний еритропоез, скорочення тривалості життя еритр. Інтрамедулярне руйнування еритрокаріоцитів, посилення гемолізу в селезінці, печінці,крові. Кровоточивість (тромбоцитопенія), інфекційні процеси (гранулоцитопенія). Збільшення в крові вмісту Fe, ціанокоболаміну, еритропоетину.
11. Залізодефіцитні анемії. Етіологія, патогенез, зміни периферичної крові. Поняття про залізорефрактерні анемії
ЗДА – анемія, зумовлена нестачею Fe в організмі насл порушення балансу між його надходженням, використанням, втратою.
Етіологія. Повторні кровотечі – втрата Fe з еритроцитами (маткові, шлунково-кишкові, ниркові, легеневі при геморагічному синдромі). Недостатнє надходження Fe з їжею, підвищені втрати в періоді росту, дозрівання о-му, під час вагітності, лактації, зниження всмоктування при хворобах травного каналу (гіпоацидний гастрит, хрон ентерит) чи резекції відділів, порушення транспорту Fe (гіпотрансферинемія – ураження печінки, спадкатрансферинемія), недостатня утилізація Fe з резервів (інфекція, інтоксикація, глистна інвазія) та його депонування (гепатит, цироз печінки).
Патогенез. Дефіцит Fe – порушення утворення гему Hb – зменшення к-ті Hb, зниження киснево-транспортної ф-ції крові, розвиток немічної гіпоксії.
Компенсаторні р-ці: 1) збільшення мобілізації Fe з депо (феритин, гемосидерин); 2) посилення абсорбції Fe в травн каналі; 3) гіперплазія еритр ростка; 4) збільш інтенсивності гліколізу.
Пат р-ції: 1) вичерпування резервів Fe (зникнення гемосидерину в макрофагах hepar, splen, зменшення феритину в hepar, м‘язах; к-ті сидеробластів в КМ); 2) зменшення у сироватці крові концFe (гіпосидеремія: 1,8-5,4 мкмоль-л, норма – 12,5-30,4 мкмоль-л), зниження р-ня феритину (нижче 12 мкг-л, норма – 12-200 мкг-л), зменш коеф насичення трансферинуFe (менше 20%, норма 30-50%) – зменшення транспорт Fe в КМ; 3) поруш надходження Fe в еритр, зниження синтезу гему; 4) неефективний гемопоез – посилення гемолізу еритрокаріоцитів у КМ, еритроцитів у крові – зменш тривалості життя еритр; 5) морф зміни в еритр КМ: гіпохромія, мікроцитоз і деструкція ядерних клітин (каріорексис, вакуолізаціяцитопл еритроцитів і нормобластів). 6) розвиток гемічної (зменш КЄК) та тканинної (зменш к-ті міоглобіну, активності залізовмісних ферментів ткан дихання) гіпоксії; 7) атроф і дистроф зміни у о-нах і тканинах (глосит, гінгівіт, карієс…, дистрофія міокарду).
Кров: К-ть Hb менша за к-ть еритроцитів. Мазок: гіпохромія, тіні еритроцитів, анулоцити, мікроцитоз, пойкілоцитоз.
Х-ка: патогенез – пов‘язана з порушенням еритропоезу, етіологія – набута, регенер здатність КМ – гіпорегенераторна, КП - гіпохромна(КП – 0,6, до 0,4), тип кровотворення – еритробластний, клініка – хронічна.
Залізорефракторна – анемія, що виникає внаслідок порушення включення Fe в гем при зниженні активності ферментів, що каталізують синтез порфіринів і гема.
Причини: 1) ген зумовлене зниження активності декарбоксилази копропорфіриногена – Е, що забезпечує 1 з кінцевих етапів синтезу гема; 2) зменшення к-ті піридоксальфосфата – акт ф-ми В6, внаслідок чого Fe не виходить з мітохондрій еритробл, не включ в гем; 3) блокада свинцем сульфгідрильних груп ферментів синтезу гема.
Зменшення акт-ті Е синтезу порфіринів і гема призводить до зниження утилізації Fe і порушення синтезу гема – недост. еритропоезу, розвгіпозромної анемії.
12. Анемії, спричинені недостатністю вітаміну В12 та/або фолієвої кислоти. Причини виникнення дефіциту вітаміну В12 і фолієвої кислоти. Злоякісна анемія Аддісона-Бірмера. Загальні порушення в організмі при дефіциті вітаміну В12 та/або фолієвої кислоти. Гематологічна характеристика В12-, фолієводефіцитних анемій.
Причини дефіциту В12
· Аліментарна недостатність
· Порушення утворення і вивільнення в слину кобалофілінів
· Недостатність пепсину при ахілії чи гіпосекреції
· Дефіцит внутрішнього фактора Кастла
· Порушення панкреатичної секреції зі зменшенням вмісту і акттвності протеоліттчних ферментів
· Порушегня функції тонкої кишки: резекція великої ділянки або хронічні проноси
· Порушення утворення транскобаламінів у печінці та транспорт їх в ентероцити
· Недостатнє депонування В12 у печінці
· Посиленне використання вітаміну В12 у вагітних
· Конкурентне використання вітаміну В12 гельмінтами і мікрофлорою кишечника
Причини дефіциту фолієвої кислоти
· Аліментарна недостатність
· Порушення всмоктування
· Посилене використоння, що не компенсується збільшенням надходження фолієвої кислоти ззовні
· Втрата вітаміну, наприклад, при гемодіалізі, при порушеннях процесів реабсорбції в нирках
Перніціозна анемія ( анемія Аддісона –Бірмера )
Найпоширеніший варіант В12 дефіцитної анемії зумовлений недостатністю внутрішнього фоктора Кастла. Вважається що в його основі лежить хронічний атрофічний гастрит, що має аутоімуне походження.
Про це свідчать такі фактори:
· Зменшення кількості паріетальних клітин, що утворюють внутрішній фактор Кастла, відбувається на тлі інфільтрації слизової оболонки шлунку лімфоцитами і плазматичними клітинами
· У плазмі та шлунковому соці часто виявляються кілька видів імуноглобулінів: антитіла, що блокують зв'язування внутрішнього фактора Кастла з вітаміном В12, і що перешкоджають взаємодії комплексу цих сполук з рецепторами ентероцитів
· Така анемія нерідко асоціцована з іншими аутоімуними хворобами, зокрема аутоімуним ураженням щитоподібної залози і мозкової речовини наднирникових залоз.
Відміність перніціозної анемії від недостатності вітаміну В12 в тому, що введення препаратів ціанокобаламіну усуває всі клінічні прояви крім хронічного атрофічного гастриту. Це вказує на те, що змвни слизової оболонки шлункує власне причиною недостатності вітаміну В12 в організмі.
Є основні три групи прояву В12- дефіцитної анемії:
1. Гематологічний синдром зумовлений панцитопенією – зменшенням кількості всіх формених елементів крові.
· Анемія та пов'язана з нею гіпоксія
· Лейкопенія, що є причиною зниженнч резистентності організму до інфекцій
· Тромбоцитопенія, яка стає причиною розвитку геморагічного синдрому
2. Ураження травного каналу, що виявляють себе розвитком запально-атрофічних змін в епітелії язика ( глосит) та слизових оболонок рота ( стоматит), стравоходу ( езофагіт), шлунку ( ахілічний гастрит), тонких кишок ( дуоденіт, ентерит)
3. Неврологічний синдром де уражаються задні і бічні стовпи спинного мозку, зумовлені дегенерацією мієліну. Це зумовлює появу рухових і сенсорних розладів.
При недостатності фолієвої кислоти не розвивається неврологічний синдром.
При В12-дефіцитній анемії в крові й червоному кістковому мозку є клітини патологічної регенерації – мегалобластів та їх без'ядерних форм- мегалоцитів.
1. Колірний покащник збільшений
2. Знижена кількість еритроцитів
3. Високий вміст гемоглобіну в еритроцитах
4. Збільшення діаметру еритроцитів ( макроцитоз, мегалоцитоз)
5. Пойкілоцитоз і анізоцитоз
6. В мегалоцитах- тільця Жолі, кільця Кебота, базофільна зернистість
7. Ретикулоцитів і поліхроматофілів мпйже нема
8. Зменшений вміст гранулоцитів і тромбоцитів
9. Гіганські нейтрофіли з гіперсегментованими ядрами в мазках
13. Гіпо- та апластична анемія. Етіологія, патогенез, прояви.
Гіпопластична ( апластична) анемія – це хвороба системи крові чи окремий синдром, що характеризується пригніченням кровотворної функції червоного кісткового мозеу та виявляє себе недостатнім утвореням еритроцитів, гранулоцитів і тромбоцитів або одних тільки еритроцитів.
Причини. Набуті:
· Фізичні факторт ( іонізуюча радіація)
· Хімічні агенти ( бензол, свинець, пари ртуті, лікарські препарати)
· Біологічні фактори ( віруси гепатиту, герпеск, цитомегаловірус)
Ідіопатична форма причинине встановоені 50-75% усіх випадків гіпопластичної анемії.
Наведені етіологічні чинники поділяються на 2 групи:
1. Фактори з облігатним ( обовязковим ) мієлотоксичним ефектом. Це іонізуюча радіація, бензол і його похідні, цитостатики та інші. Ушкоджувальна дія їх на червоний кістковий мозок залежить від дози і тривалості. У більшості випадків кровотворення відновлюється після припинення впливу.
2. Фактори з факультативним мієлотоксичним впливом.
Спадково зумовленої форми гіпопластичної анемії є анемія Фанконі. Сутність дефекту полягає в порушенні систем репарації ДНК.
При гіпопластичній анемії уражуються стовбурові клітини червоного кісткового мозку, а доказом цього є панцитопенія.
Механізми ураження стовбурових клітин:
1. Необоротне ушкодження ДНК, яке запускає механізми програмованої загибелі клітин – апоптоз. Вважають, що в такий спосіб діють фактори з облігатним, дозозолежним мієлотоксичним ефектом.
2. Імунне ушкодження клітин. Виникає у разі появи на поверхні стовбурових клітин нових антигенів, до яких немає імунологічної толерантності. Причиною цього стають спонтанні або індуковані мієлотоксичними факторами мутації. Експресія додаткових антигенів стпє причиною імунних реакцій, спрямованих на знищення стовбурових клітин.
Результатом загибелі стовбурових клітин стає зменшення їхньої кількості, що веде до істотного зменшення пулів проліферуючих і дозріваючих клітин – червоний кістковий мозок спустошується та заміщується жировою тканиною, що називають аплазією.
Прояви:
1. Зменшення кількості еритроцитів, концентрації гемоглобіну
2. Колірний показник в межах норми
3. У мазку крові невиявляються регенераторні форми еритроцитів
4. Зменшення вмісту тромбоцитів і гранулоцитів
Виокремлюють 3 основні клінічні синдроми:
1. Анемія і пов'язаний з нею гіпоксичний синдром
2. Запальні процеси, зумовленні інфекційними агентами
3. Геморагічний синдром
14.Лейкоцитоз, принципи класифікації. Причини та механізми розвитку реактивного та перерозподільного лейкоцитозу (на прикладі зсувів після прийому їжі).
Класифікація:
1. В залежності від причин:
А) фізіологічний:
· Емоційний лейкоцитоз – виникає під час сильних емоцій
· Міогений виникає під час інтенсивних фізичної роботи
· Статичний – характерний для переходу людини з горизонтального в вертикальне положення
· Аліментарний – виникає під час пртйому їжі
· Лейкоцитоз вагітних
· Лейкоцитоз новонароджених
Б) патологічний
· Інфекційні хвароби
· Запальні та алергічні процеси
2. Лейкоцитоз може бути абсолютним або відносним. Для абсолютного лейкоцитозу характерне збільшення абсолютної кількостілейкоцитів в одиниці об'єму крові. Про відносний лейкоцитоз ідеться в тому випадку, коли зростає відносний вміст окремих формених елементів у периферійній крові.
3. За механізмом розвитку:
· Реактивний
· Перерозподілений
· Пухлинного походження
4. В залежності від виду лейкоцитів;
· Нейтрофільний лейкоцитоз ( нейтрофільоз). Характерний для:
А)гнійно-запальних процесів спричинений гноетворними бактеріями
Б) асептичного запалення, зумовленого некрозом тканин
В) впжкого кисневого голодування
Г) ендогенної інтоксикації (уремії)
· Еозинофільний лейкоцитоз ( еозинофілія)
А) алергічних реакціях І типу
Б) паразитарних інфекціях в тому чисді гельмінтози
В)деякі злоякісні пухдини, зокрема хронічний мієлолейкоз
· Базофільний лейкоцитоз
А) хронічний мієлолейкоз
Б) гемофілія
В) хвороби Вакеза ( поліцитемії)
· Лімфоцитарний лейкоцитоз ( лімфоцитоз)
А)деяких гострих вірусних інфекціях ( вірусний гепатит,цитомегаловірусна та герпевірусна інфекції)
Б) хронічних інфекційних хвороб, що супроводжуються тривалою імуною стимуляцією ( туберкульоз, сифіліз, бруцельоз)
В) хронічний лімфолейкоз
· Моноцитарний лейкоцитоз ( моноцитоз)
А) деяких хронічних інфекціях ( туберкульоз, бруцельоз)
Б) ігфекційному мононуклеозі
В) інфекції збудником яких є рикеції і найпростіші ( висипний тиф, малярія)
Механізми розвитку лейкоцитозів:
1. Перехід лейкоцитів із червоного кісткового мозку у кров. Цей механізм лежить у основі реактивного лейкоцитозу, що виникає як реакція червоного кісткового мозку на патогенні впливи. Він закономірно розвивається при інфекційних захворюваннях, запаленні, дії низьких доз токсичних речовин.
Реактивний лейкоцитоз зумовлюється посилегням лейкопоезу і збільшенням переходу лейкоцитів з резервного пулу червоного кісткового мозку в кров.
· Посилення лейкопоезу має в своїй основі дію на кровотворні клітини ІІ класу ( лейко поетини). До цієї групи відносять цитокіни та фактори росту.
З цитокінами повязують стимуляцію лейкопоезу. Фактори росту посилюють розмноження і деференціацію кровотворних клітин називають колонієстимулюючим фактором.
У вогнищі запалення відбувається активація макрофагів, вони виділяють в тканини і кров цитокіни, які діють на клітини строми червоного кісткового мозку. Т-лімфоцити і клітини ендотелію судин, зумовлюють виділення кількох факторів росту: гранулоцит-моноцитарного, гранулоцитарного, моноцитарного колоніє стимулюючих факторів. Вплив цих факторів накровотворні клітини, запрограмовані на шлях міопоетичного розвитку, спричиняється до посилення поділу їх і диференціацію в клітин гранулоцитарного ряду й моноцити.
· Збільшення переходу лейкоцитів із резервного пулу червоного квсткового мозку в кров є другим механізмом реактивного лейкоцитозу. Цілий ряд прозапальних цитокінів, що посилено продукуються при гострому запаленні, збільшують проникність епітелію судин червоного кісткового мозку й у такий спосіб значно полегшують вихід зрілих лейкоцитів та безпосередніх їхніх попередників у периферичну кров.
2.Перерозподіл лейкоцитів крові. Суть цього механізму полягає в тому, що лейкоцити переходять із пристінкового пулу в циркулюючий, збільшуючи в такий спосіб кількість лейкоцитів в одиниці об'єму крові.
Більшість форм фізіологічного лейкоцитозу має в своїй основі перерозподільний механвзм. Його особливостями є:
· Короткочасний характер із швидким поверненням кількості лейкоцитів до норми після завершення дії причини.
· Збереження нормального кількісного співвідношення різних видів лейкоцитів
· Відсутність дегенеративних змін лейкоцитів
15.Механізм фізіологічного лейкоцитозу новонароджених. Особливості лейкоформули в дітей різного віку
Картина білої крові у дітей з часом змінюється, і навіть в одному і тому ж віці кількість білих кров'яних тілець може коливатися у дуже широких межах. Так в новонароджених в перші 8-12 години життя кількість лейкоцитів складає в середньому 20•10 9/л з коливаннями (9-12)•10 9/л. У перші дні життя дитини спостерігається нейтрофільний лейкоцитоз із зрушенням вліво. Число нейтрофілів складає 65-66 % від загальної кількості лейкоцитів, тоді як кількість лімфоцитів –16-34 %.
До 5-6-го дня відсотковий вміст нейтрофілів і лімфоцитів зрівнюється, що розцінюється як «перше перехрещення» в зміні їх кількісних співвідношень. До кінця 1-го місяця життя число нейтрофілів зменшується до 25-30 %, а лімфоцитів зростає до 55-60 %. У віці 4-5 років рівень лімфоцитів зменшується, а нейтрофілів зростає і в період між 5-м і 6-м роками зрівнюється («друге перехрещення»). До 12-14 років встановлюються такі ж відсотковеспіввідношення між цими формами, як і у дорослих.
Відносний і абсолютний нейтрофільоз в перші дні періоду новонародженості пояснюється потраплянням в організм дитяти через плаценту материнських гормонів (гормональна теорія), а також згущуванням крові в перші години внутріутробного життя, розсмоктуванням внутрітканевих крововиливів, всмоктуванням продуктів розпаду тканин самої дитини у зв'язку з недостатнім поступленям їжі в перші дні життя.
16.Нейтрофільний, еозинофільний, базофільний, лімфоцитарний і моноцитарний лейкоцитоз (при яких патологічних процесах та захворюваннях виникає кожен з цих видів).
· Нейтрофільний лейкоцитоз ( нейтрофільоз). Характерний для:
А) гнійно-запальних процесів спричинений гноетворними бактеріями ( абсцесс, флегмона, сепсис)
Б) асептичного запалення, зумовленого некрозом тканин ( інфаркт міокарда, опіки)
В) важкого кисневого голодування ( гострий гемоліз, велика гостра крововтрата)
Г) ендогенної інтоксикації (уремії)
· Еозинофільний лейкоцитоз ( еозинофілія)
А) алергічних реакціях І типу ( анафілактичних)
Б) паразитарних інфекціях в тому чисді гельмінтози
В)деякі злоякісні пухдини, зокрема хронічний мієлолейкоз
· Базофільний лейкоцитоз
А) хронічний мієлолейкоз
Б) гемофілія
В) хвороби Вакеза ( поліцитемії)
· Лімфоцитарний лейкоцитоз ( лімфоцитоз)
А)деяких гострих вірусних інфекціях ( вірусний гепатит,цитомегаловірусна та герпевірусна інфекції)
Б) хронічних інфекційних хвороб, що супроводжуються тривалою імуною стимуляцією ( туберкульоз, сифіліз, бруцельоз)
В) хронічний лімфолейкоз
· Моноцитарний лейкоцитоз ( моноцитоз)
А) деяких хронічних інфекціях ( туберкульоз, бруцельоз)
Б) ігфекційному мононуклеозі
В) інфекції збудником яких є рикеції і найпростіші ( висипний тиф, малярія)
17. Місцева і загальна дія на організм іонізуючого випромінювання. Гостра променева хвороба, її форми. Патогенез кістково-мозкової форми гострої променевої хвороби. Віддалені наслідки дії іонізуючого опромінення.
Опромінення може бути внутрішнім, при проникненні радіоактивного ізотопа всередину організму, та зовнішнім; загальним (опромінення всього організму) та місцевим; хронічним ( при дії протягом тривалого часу) та гострим (одноразовий, короткочасний вплив).
Гостра променева хвороба (ГПХ) – група синдромів, які розвиваються після короткочасного опромінення всього тіла або більшої його частини в дозі понад 1 Гр. та при обов’язковій наявності ознак пригнічення кровотворення і обмеженні часу реалізації патологічних зрушень строком 2-3 місяці.
Залежно від поглиненої дози опромінення виділяють три форми гострої променевої хвороби: кістково-мозкову (поглинена доза — від 0,5 до 10 Гр), кишкову (від 10 до 50 Гр) і мозкову (від 50 до 200 Гр). У клініці кістково-мозкової форми розрізняють 4 періоди: 1) період первинних реакцій (тривалість - 1—2 доби); 2) період удаваного благополуччя (кілька діб); 3) період виражених клінічних ознак; 4) завершення.
Типова або кістковомозкова форма ГПХ, при якій основним патогенетичним механізмом розвитку захворювання є ураження органів кровотворення, супроводжується виникненням провідного клінічного прояву — синдрому пангемоцитопенії і тісно пов’язаних з нею синдромів кровоточивості та інфекційних ускладнень.
Віддалені наслідки опромінення - соматичні та стохастичні ефекти, які проявляються через тривалий час (кілька місяців або років) після одноразового або в результаті хронічного опромінення.
Включають в себе:
-зміни в статевій системі;
-склеротичні процеси (заміна паренхіми органів щільною сполучною тканиною; не є самостійним захворюванням, а служить патоморфологическим проявом іншого основного захворювання);
-променеву катаракту (офтальмологічне захворювання, пов'язане з помутнінням кришталика ока і яка викликає різні ступені розладу зору аж до повної його втрати);
-імунні хвороби;
-радіоканцерогенез (злоякісна пухлина або пухлина, властивості якої найчастіше роблять її вкрай небезпечною для життя організму);
-скорочення тривалості життя;
-генетичні та тератогенні ефекти (виникнення вад розвитку і каліцтв внаслідок опромінення, як правило, проявляються у наступних поколінь).
18. Патогенний вплив надмірної та недостатньої інсоляції ультрафіолетовими променями. Фотосенсибілізація.
Біологічна дія УФ-випромінювання сонячного світла проявляється, перш за все, у його позитивному впливі на організм людини. УФ-опромінення є життєво необхідним чинником. Відомо, що при тривалій недостатності сонячного світла, виникають порушення фізіологічної рівноваги організму, розвивається симптомокомплекс, що має назву "світлове голодування".
Найбільш частими наслідками недостатності і сонячного світла є послаблення захисних імунобіологічних реакцій організму, загострення хронічних захворювань, функціональні розладнання нервової системи. Це стосується робітників, що працюють у шахтах або у безліхтарних і безвіконних цехах і на тих об’єктах, що не мають природного освітлення (метрополітен) і т. ін.
Великi дози iонiзуючого випромінювання за певних умов здатнi спричинити злоякiсний рiст, променеву хворобу, злоякiсне бiлокрiв’я.Найчутливiші до iонiзуючого опромінення кровотворна, лiмфоїдна та епiтелiальнi тканини i особливо епiтелiй залоз системи травлення, статевої сфери, шкiри, ендотелiй судин.
Фотосенсибілізація — це підвищення чутливості організму до дії ультрафіолетового випромінювання. Речовини, що викликають ефект фотосенсибілізації, отримали назву фотосенсибілізаторів. Розрізняють екзогенні (еозин, риванол, акридин, хінін, сульфаніламіди) і ендогенні фотосенсибілізатори (жовчні кислоти, гематопорфірини, холестерол, білірубін). Вважають, що під впливом фотосенсибілізаторів ультрафіолетові промені починають взаємодіяти з тими молекулами тканин, з якими під час відсутності фотосенсибілізаторів вони не взаємодіють. Крім того, є підстави думати, що в цих умовах прискорюється проходження енергії ультрафіолетових променів по вуглецевих скелетах біологічних молекул.
19. Дія на організм високого та низького атмосферного тиску. Патогенез синдромів компресії і декомпресії. Вибухова декомпресія.
Порушення, що зумовлені дією на організм низького атмосферного тиску, можуть виникати в людини при підйомі у гори, на літальних апаратах без кисневих приладів. Особливого значення цей фактор набуває за умов висотних польотів і в космосі при аварійних ситуаціях. В умовах гіпобарії відбуваються наступні зміни фізико-хімічних властивостей газів та рідин:
зниження парціальних тисків газів повітря (при відсутності змін їх процентного співвідношення);
збільшення об’єму газів;
зниження розчинності газів (десатурація);
зниження температури закипання рідин.
Кожен з цих факторів спричинює низку причинно-наслідкових змін, які спостерігаються при гіпобарії, але провідне місце належить падінню парціального тиску кисню (рО2), що вперше було доведено в дослідах Поля Бера у 1878 році. Так, на рівні моря рО2 дорівнює 160 мм рт. ст., а на висоті 12000 м – 30 мм рт. ст. Людина при таких низьких показниках парціального тиску кисню зберігає свідомість не довше 30-40 секунд.
Нозологічними формами, що виникають під впливом зниження атмосферного тиску є гірська і висотна хвороби.
Людина перебуває в умовах підвищеного тиску газового і водного середовища в процесі водолазних спусків і кесонних робіт, під час пірнання і підводного плавання з аквалангом, при лікуванні штучними газовими сумішами або киснем в камерах підвищеного тиску і бароопераційних, при порятунку з відсіків затонулого підводного човна. Свої особливості має праця водолазів, акванавтів, а також підводників, які за родом діяльності систематично або періодично піддаються дії підвищеного тиску газового і водного середовища. Підвищення атмосферного тиску супроводжується наступними змінами фізико-хімічних властивостей газів:
підвищення парціальних тисків газів у газовій суміші;
підвищення розчинності газів (сатурація);
зменшення об’єму газів.
Провідну роль в розвитку гіпербаричного стану відіграє зміна розчинності газів.
Сатурація (лат. saturatio – насичення) – насичення рідин і тканин організму газами за умов підвищеного барометричного тиску.
Провідні ланки патогенезу: азотне отруєння, кисневе отруєння, баротравма.
Хвороба декомпресії виникає при швидкому поверненні людини в умови нормального атмосферного тиску після водолазних робіт, робіт у кесонах (кесонна хвороба). При цьому розчинені в крові й тканинах гази (азот, кисень) у великій кількості переходять у газоподібний стан, утворюючи величезну кількість бульбашок, - відбувається десатурація.
Бульбашки газів, затримуючись у крові й тканинах, можуть закупорювати кровоносні судини, справляючи тиск на клітини, подразнюючи рецептори (газова емболія). Клінічна картина такої хвороби визначається локалізацією газових бульбашок. Найчастіше виникає біль у суглобах, свербіж шкіри; у важких випадках - порушення зору, параліч, втрата свідомості. Щоб уникнути подібних порушень, декомпресію варто проводити повільно, щоб швидкість утворення газів не перевищувала можливості легень щодо їх виведення. Вибухова декомпресія виникає у випадку швидкого перепаду атмосферного тиску від нормального до зниженого, що буває при розгерметизації висотних літальних апаратів (літаків, космічних кораблів). У розвитку цього синдрому має значення баротравма легень, серця й великих судин внаслідок різкого підвищення внутрішньо-легеневого тиску. Розрив альвеол і судин сприяє проникненню газових бульбашок у кровоносну систему (газова емболія). У крайніх випадках настає миттєва смерть унаслідок закипання крові й інших рідин організму, а також у результаті блискавичної форми гіпоксії.
20. Хімічні патогенні чинники як проблема екології і медицини. Токсичність, канцерогенність, тератогенність хімічних сполук. Екзоінтоксикації. Патофізіологічні аспекти паління, алкоголізму і наркоманії. Значення шкідливих звичок у дитячому віці.
Особливу небезпеку становлять хімічні речовини, які залежно від їх практичного використання можна поділити на:
• промислові отрути, які використовуються у виробництві (розчинники, барвники) є джерелом небезпеки гострих і хронічних інтоксикацій при порушенні правил техніки безпеки (наприклад, ртуть, свинець, ароматичні сполуки тощо);
• отрутохімікати, що використовуються у сільському господарстві для боротьби з бур’янами та гризунами (гербіциди, пестициди);
• лікарські препарати;
• хімічні речовини побуту, які використовуються як харчові добавки, засоби санітарії, особистої гігієни, косметичні засоби;
• хімічна зброя.
Токсичність – це здатність речовин шкідливо впливати на життєдіяльність організмів
Токсичні речовини — це речовини, які викликають отруєння усього організму людини або впливають на окремі системи людського організму (наприклад, на кровотворення, центральну нервову систему).
Ці речовини можуть викликати патологічні зміни певних органів, наприклад, нирок, печінки. До таких речовин належать такі сполуки, як чадний газ, селітра, концентровані розчини кислот чи лугів тощо.
Канцерогенність – чинники навколишнього середовища, які впливаючи на організм підвищують вірогідність виникнення злоякісних пухлин.
Канцерогенні речовини викликають, як правило, злоякісні новоутворення — пухлини (ароматичні вуглеводні, циклічні аміни, азбест, нікель, хром тощо).
Тератогенність - чинники навколишнього середовища, які спричиняють порушення розвитку організму.
Тератогенним вважається хімічний, фізичний або біологічний чинник, що відповідає таким критеріям: 1) доведено зв’язок між дією чинника і формуванням дефекту розвитку; 2) епідеміологічні дані підтверджують цей зв’язок; 3) дія ушкоджувального чинника збігається з критичними періодами внутрішньоутробного розвитку; 4) при нечастому впливі ушкоджувального фактора характерні дефекти розвитку формуються рідко. Основні групи Т.ч. включають ЛП і хімічні речовини, іонізуюче випромінювання, інфекції, метаболічні порушення і шкідливі звички вагітної.
Екзоінтоксикації – поступають в організм з навколишнього середовища.
Екзогенні отрути діляться на неорганічні (кислоти, луги, солі свинцю, ртуті, миш'як)) і органічні (алкоголь, ефір, хлороформ, ціанисті сполуки). У свою чергу органічні отрути діляться на отрути рослинного походження (алкалоїди, глюкозиди) і тваринного (зміїна отрута). Отрути можна класифікувати в залежності від переважної дії їх на окремі органи і системи (кров'яні отрути - бертолетова сіль, окис вуглецю; печінкові отрути - флоридзин; нервові - миш'як, стрихнін та ін.).
Алкоголізм, пат аспекти
Алкоголізм – захворювання, різновид токсикоманії , характеризується залежністю до алкоголю ( етиловому спирту) , з психічною і фізичною залежністю від нього.
В патогенезі виділяють по Стрельчуку три стадії:
ü Компенсуюча – головний симптом – необмежена тяга до вживання алкоголя, втрата « відчуття міри» по відношенню до випитого, формування толерантності до алкоголю, і легкої форми обстинентного синдрома.
ü Наркоманна – хвороблива залежність до алкоголю збільшується. Зростання психічних змін : концентрація всіх інтересів на алкоголь, егоцентризм – кінцева форма індивідуалізма, і егоїзма, притуплення відчуття долгу, і інших вищих емоцій. Кінцева форма обстинтного синдрома.
ü Термінальна - ознаки психічного не врівноваження , падіння толерантності до алкоголю.
Ефекти дії на ЦНС:
Ø Фаза збудження – характеризується ейфорією , відчуттям бодрості, і приплива сил. Під час цієї фази порушується метаболізм нейронів кори головного мозку, зменшується кількість серотоніну, збільшується виділення адреналіну, норадреналіну,
Ø Фаза пригнічення – дисфория , зменшується виділення адреналіна, норадреналіна, депресія.
Порушення роботи печінки:
Цироз печінки – тяжке захворювання печінки, супроводжуючу незворотнім заміщенням, паренхіматозної тканини, печінки фіброзною, сполучною тканиною або стромою. Циротична печінка збільшена, бугриста, шерхувата. Частіше всього цироз розвивається при довготривалій інтоксикації алкоголем. Важливий фактор – некроз гепатоцитів, обосновлений прямою токсичною дієюалкоголя, а також аутоімунними процесами.
Пат аспекти наркоманії
Наркотики – природні речовини, які можуть визвати наркоманію.
Характеристика наркотиків: діють на нервову систему; мають негативні соціальні наслідки;
Види наркоманії:
V Каннабізм – приймання препаратів канна біса( висушені жіночі квіти) – гашиша, марихуани;
Визиває відчуття релаксації, порушення концентрації уваги,
v Кокаинизм - вживанні кокаїну, використовується у вигляді білого порошка.
v Опійна наркоманія – використанні опіатів – морфіна, кодеіна, промедол,
Ефекти: зниження гостроти больових відчуттів, седативний ефект, ейфорія, угнетення дихання, тошнота, рвота, порушення моторики ЖКТ, зниження секреторної її функції.
Значення шкідливих звичок у дитячому віці.
Алкоголь. В організмі дитини чи підлітка алкоголь перш за все проникає в кров, печінку, мозок. У зв'язку з незрілістю центральної нервової системи, вона найбільш вразлива для дії етанолу. Результатом такої дії є зміна особистості дитини, порушується:
логічне абстрактне мислення;
інтелект;
пам'ять;
емоційне реагування.
При впливі алкоголю уражаються практично всі системи організму підлітка. Згідно зі статистикою, 5-7% отруєнь у дітей припадає на частку алкогольних інтоксикацій. Явища сп'яніння у дітей і підлітків розвиваються швидко і можуть завершитися комою. Артеріальний тиск і температура тіла підвищуються, рівень глюкози в крові, кількість лейкоцитів падає. Короткочасне збудження, викликане прийомом алкоголю, швидко переходить в глибокий інтоксикаційний сон, нерідкі судоми, навіть летальний результат. Іноді реєструють психічні порушення з маренням і галюцинаціями.
Наркотики. Багато наркотиків хімічно подібні нейротрансміттерам - речовин, які виділяються нервовими закінченнями при стимулюючому імпульсі. Нейротрансмітери взаємодіють з рецепторами - сенсорними нервовими закінченнями, які можуть приймати імпульси і реагувати на них. До числа нейротрансмітерів відносяться серотонін і ендорфіни. Вони контролюють настрій, емоції і гормональну функцію, а також пригнічують біль.
Куріння. При курінні нікотин викликає звуження артерій, ускладнюючи тим самим доступ кисню, що переноситься кров'ю, до серця, мозку, нижніх кінцівок і іншим органам. Причому нікотин діє швидко, звужуючи судини протягом хвилини після першої затяжки. Нормальний просвіт судин відновлюється нескоро. Тому нерідко курець постійно тримає свої кровоносні судини в спазматичному стані.
Крім того, нікотин, потрапляючи в кров, подразнює наднирники, які викидають в кров гормон адреналін, теж звужує судини і підвищує кров'яний тиск. Постійне надходження в кров адреналіну викликає стійкі зміни судинних стінок.
Для дії нікотину характерно також зниження апетиту, порушення функцій шлунка, різке зниження насиченості організму вітаміном С, не повернуться до нормальних навіть при введенні великих доз аскорбінової кислоти.
Нікотин надає згубну дію на статеву функцію,вагітність, на зір і слух.
21. Інфекційний процес, загальні закономірності розвитку. Механізми захисту організму від інфекції.
Інфекційний процес - типовий патологічний процес, що лежить в основі розвитку інфекційних хвороб (ІХ)
Етіологія інфекційного процесу
Серед інфекційних агентів розрізняють наступні:
віруси
рикетсії
бактерії
гриби
найпростіші
гельмінти
Розрізняють облігатно-патогенні та умовно-патогенні мікроорганізми. Потрапляння останніх у організм викликає захворювання лише за певних умов: велика кількість життєздатних збудників, знижена стійкість організму і т. д.
Важливе значення мають умови навколишнього середовища, які здатні впливати одночасно на макро- і мікроорганізм. До несприятливих умов для мікроорганізмів належать: зміна температури, висушування, радіацію, дезинфікуючі засоби. На реактивність макроорганізму негативно впливають вологість, низька температура та соціальні фактори.
Механізми захисту організму від інфекцій
Механізми захисту від інфекцій можуть бути поділені на специфічні і неспецифічні.
Специфічні механізми спрямовані проти певного виду збудника, неспецифічні – проти багатьох чи усіх.
Специфічні механізми.
Внаслідок потрапляння збудника у організмі формується імунна відповідь. Яка ланка імунної відповіді буде включена залежить від особливостей збудника та вхідних воріт інфекції. Якщо мікроорганізми розмножуються позаклітинно (бактерії) – включається гуморальна ланка, якщо внутрішньоклітинно (гриби, віруси) – клітинна ланка імунітету. Крім цього, у відповідь на дію екзотоксинів у організмі утворюються антитоксини, що нейтралізують їх (при первинному потраплянні збудника часто із запізненням, тому не завжди є ефективними).
Віруси при розмноженні в місці потрапляння у організм ініціюють механізми місцевого імунітету (синтез IgA).
Завдяки утворенню клітин імунологічної пам’яті (лімфоцитів), після перенесеного інфекційного процесу розвивається природний активний імунітет. Однак, в одних випадках він є пожиттєвим, а в інших – на короткий термін.
Неспецифічні механізми
Неспецифічні механізми захисту організму від інфекційних агентів включаються на різних рівнях:
системному
органному
тканинному
гуморальному
клітинному
До системних механізмів належать рефлекторні реакції, перерозподіл кровоплину, зміна ендокринної регуляції, підсилення функцій видільної системи, підсилення антитоксичної функції печінки.
До органних і тканинних механізмів належать бар’єрні функції шкіри і слизових, секреторні процеси, скупчення лімфоїдної тканини.
До гуморальних і клітинних механізмів належать активація системи інтерферону, дія лізоциму і інших гуморальних факторів неспецифічної резистентності організму, активація фагоцитозу.
22. Патогенез гострих і хронічних лейкозів. Картина крові при гострих та хронічних лейкозах; системніпорушення в організмі при лейкозах.
Лейкоз – пухлина, що виникає з кровотворних клітин КМ.
Гострий – злояк пухлина, що складається з бластних клітин І,ІІ,ІІІ,ІV класів гемопоезу, що втратили здатність до дозрівання.
Хронічний – пухлина, що складається переважно з дозріваючих і зрілих клітин V,VI класів, оскільки основна маса лейкозних клітин диференціюється до зрілих форм.
Головна риса патогенезу гострих лейкозів – лейк. клітини, отримавши здатність до неконтрольованого беззупинного поділу, повністю втратили здатність дозрівати (диференціюватись).
Відбувається пухлинна трансформація кровотворних клітин, гол. чином СКК, НСКК, клітин-попередників лімфо-, мфєлопоезу. Наслідок – виникнення пухлин.
Характерне для лейкозу первинне утворення непластичного клону в КМ.
Кров. Гострі. 1) мієлобластний: мієлобласти, метамієлоцити, паличкояд, сегментоядл-ц, відсутні перехідні ф-мил-ц, нема промієлоцитів, мієлоцитів (лейкемічний провал). 2) лімфобластний: лімфобласти, норм клітини крові.
Хронічні. 1) мієлоцитарний: метамієлоцити, промієлоцити, мієлоцити, паличкояд, сегментоядл-ц. 2) лімфоцитарний: 80-90% лімфоцитів, про лімфоцити, тіні Гумпрехта (напівзруйн ядра лімфоцитів).
23. Прояви пухлинноїпрогресії при лейкозах, поняття про моноклонову та поліклоновустадіюлейкозів, «бластний криз», «лейкемоїдний провал», лейкозніінфільтрати.
Пухлинна прогресія - лейкозні клітини набувають велику злоякісність. Вони стають морфологічно і цитохімічних недиференційними, в кровотворних органах і крові збільшується кількість бластних клітин з дегенеративними змінами ядра і цитоплазми. Лейкозні клітини поширюються за межі кровотворних органів, утворюючи лейкозні інфільтрати в самих різних органах. Внаслідок відбору знищуються клітини тих клонів, на які діяла імунна система і гормони організму, цитостатичні засоби. Домінують клони пухлинних клітин, найбільш стійких до цих впливів.
Моноклонова (відносно доброяк). 1) акт проліферуючі кл утвореного лейкозного клону відзначаються однаковими гено-, фенотипом, однаковими маркерами (АГ, хромосомні, б-х), при хронічній формі – схожі з норм кл крові, при гострому можлива цитохім та імунологічна ідентифікація бластних клітин. 2) відсутність чи знижена здатність лейкозних кл до дозрівання, це зумовлює накопичення пухл клітин, що акт проліферують.
Поліклонова (злояк). Замінює моноклоновувнаслпухл прогресії. 1) виникнення спонтанної та індукованої мутації внасл нестабільності геномів пухл кл. Утворення кількох нових клонів. Пухлина – фено-, генотипово неоднорідна. 2) лейк кл стають злоякісними, морф і цитохімнедиференц, втрачають ф-ції. Збільш к-тьбластних кл (бластний криз), 3) здійснення добору, внасл якого знищ кл тих клонів, на які діяли ф-ри імунного захисту, гормони, цитостатичні засоби. Домінування клонів, найб резистентних до впливів до цих ф-рів. 4) витіснення інтенспроліферуючихлейк кл в КМ норм кровотв кл – пригнічення норм кровотворення та імунної сс.-ми, розв анемічного, гемораг, імунодеф станів. 5) поширення лейк кл за межі с-ми крові з утвлейк інфільтратів у різних о-нах.
Лейкемоїдний(лейкемічний)провал- гематологічний феномен, який характеризується наявністю у крові бластних (недозрілих) та дорослих клітинних форм і відсутністю клітин середнього рівня розвитку. Зустрічається при гострих лейкозах.
Метастазування при лейкозі супроводжується появою лейкозних інфільтратів у різних органах — печінці, селезінці, лімфатичних вузлах та ін. В органах можутьрозвиватисязміни, зумовлені обтурацією судинпухлиннимиклітинами — інфаркти, виразково-некротичніускладнення.
24. Лейкози у дітей: найбільш часті форми, клінічні прояви, патогенетичне обґрунтування можливих напрямків лікування.
Лейкози посідають ведуче місце у структурі онкологічних захворювань у дітей і складають приблизно 30 % всіх злоякісних новоутворень дитячого віку. На гострулімфобластнулейкемію (ГЛЛ) перепадає 80 %, на гострумієлобластнулейкемію (ГМЛ) – 15–20 %. Хронічнамієлобластналейкемія у дітейзустрічається с частотою 3–5 %.
ГЛ в основному обумовленізаміщеннямнормальноїгемопоетичноїтканинипухлиннимиклітинами. Таким чином, на певномуетапірозвитку хвороби в периферичнійкровіслідочікуватианемію, тромбоцитопенію, нейтропенію з формуваннямвідповіднихклінічнихсиндромів: 1) Анемічний синдром 2) Геморагічний синдром 3) Інфекційний синдром 4) Гіперпластичний 5) Інтоксикаційний синдром 6) Артралгічний (оссалгічний) синдром.
Лікування визначається клініко-гематологічною проявами і стадією захворювання. У розгорнутої стадії ХМЛ(хронічний мієлолейкоз) використовують монохіміотерапію (літалір, мієлосан). Основна мета лікування в цій стадії захворювання - зменшення маси пулу пухлинних клітин, що проліферують.
25.Механізмитромбоцитарно-судинного та коагуляційного гемостазу. Можливімеханізмипорушень.
Судинно-тромбоцитарний гемостаз–цесукупністьсудинних та клітинних (тромбоцитарних) реакцій, якізабезпечуютьзакриттяпошкоджень в стінцісудинтромбоцитарним тромбом і зупинкукровотечі із судинмікроциркуляторного русла (прекапіляри, капіляри, посткапіляри), тобтосудин з низькоюлінійноюшвидкістю кровотоку та низькимтиском.
Механізм
СТГ:1.Рефлекторний спазм судин- у
відповідь на подразнення їх стінок (подальше звуження судин пов'язане з дією
судинозвужуючих гуморальних факторів - серотонін, тромбоксан, адреналін які
виділяють тромбоцити). Цяреакція (декількахвилин), спрямована на
тимчасовузупинкукровотечіабочастковезменшення крововтрати.2.Адгезія
(прилипання) тромбоцитівдо пошкодженої стінки судини.3.Агрегація
(злипання)тромбоцитів.Дана фаза поділяється на 2 підфази:-зворотня - процес утворення конгломерату (скупчення) по 10 - 20
тромбоцитів, що пропускає через себе плазму крові. Утворюється рухлий білий
тромб і через 1 – 3хвилини закриває ушкодження.-незворотня - процес, головним
стимулятором якого є тромбін: тромбоцити стають сферичними, утворюють
відростки, що полегшує їх агрегацію, і відбувається злиття змінених тромбоцитів
в гомогенну масу, яка не пропускає плазму крові,.4.Ретракція
тромбоцитарноготромба,його ущільнення і закріплення в
пошкоджених судинах за рахунок скорочення актоміозиноподібного білка
тромбоцитів -тромбостеніну, що забезпечує віджимання та ущільнення тромбу,
зменшення згустка.
В результаті описаних процесів утворюється білий тромбоцитарний тромб, який може забезпечити зупинку кровотечі із судин мікроциркуляторного русла, але не може зупинити кровотечу з крупних судин (з великою лінійною швидкістю руху крові, з високим тиском —там такий тромб руйнується через недостатню механічну міцність).
Коагуляційний гемостаз - процесзсіданнякрові, тобтозмінаїї агрегатного стану -перехід з рідкого стану в желеподібний. В результаті таких змінутворюєтьсяфібриновийзгусток - тромб, щозакриваєотвір (пошкодження) у судині. КГЗ забезпечуєзупинкукровотечі з великих судин, де високалінійнашвидкість кровотоку і високийтиск. КГЗ протікаєу три фази, яківзаємопов'язані та взаємозалежні.
Механізм:1.Утворення кров’яної та тканинноїпротромбіназ. 2.Перетворення протромбінуутромбін.3.Перетворення фібриногену у фібрин.
Можливі механізми порушень?: Взаємний зв'язок фаз полягає в тому, що продукт попередньої реакції ініціює наступну, тобто згортання крові - це ланцюгова ферментативна реакція. У здоровомуорганізміфакторизгортаннякровізнаходяться у неактивному стані.
26.Методивизначення гемостазу. Тромбоеластографія: методика визначення, прояви гіпер- та гіпокоагуляції.
Коагулограма— метод лабораторного дослідженнякрові з метою встановлення стану згортальноїсистеми.
Методом Лі-Уайта, кров швидкозабирається в 3 силіконовіпробірки. Обсягкрові — 1 мл в кожнупробірку. Кров нагрівають до 37 ° С і нахиляють. Як тільки при нахилі кров перестаєрухатисяприрусіпробірки, процесвважаєтьсязакінченим. Час згортання по Лі-Уайту становить 4-6 хвилин.
Тромбоеластограма (еластограма, тромбоеластографія (ТЕГ))
Тромбоеластографія (ТЕГ) - метод графічної реєстрації процесів згортання крові і фібринолізу.
Тромбоеластографія базується на вимірюванні фізичної міцності згустку. Невелика кількість крові (зазвичай близько 0,4 мл) міститься в кювету, яка здійснює повільні (6 разів на хвилину) обертальні коливання на кілька (4,5º) градусів. В камеру поміщається стрижень датчика і додаються активатори згортання. Коли в кюветі формується згусток, стрижень починає обертатися разом зі згустком. Амплітуда відхилення стрижня реєструється як функція часу. На типовою кривої тромбоеластографії можна виділити різноманітні параметри, що характеризують як швидкість формування згустку, так і його фінальні характеристики.
Гіпокоагуляція - це зниження здатності крові згортатися з появою схильності до повторних кровотеч і крововиливів (спонтанним або після незначних травм).
Кровотечі можуть мати на шкірі вид точок або невеликих висипань. Крововилив може відбуватися не тільки в підшкірному просторі, але і в м'язах, а також суглобах. В основі зниження згортання крові лежать такі механізми: 1) зниження концентрації в крові прокоагулянтов; 2) недостатня активація прокоагулянтов; 3) підвищена концентрація або надмірна активація антикоагулянтів; 4) підвищена концентрація або надмірне посилення активності фібринолітичних факторів.
Гиперкоагуляційний синдром (ГКС) окреслена коагулопатия, що характеризується підвищеною готовністю до тромбозу, з клініко-лабораторними ознаками гіперкоагуляції і активації факторів згортання крові, але без наявності гострого тромбозу. ГКС супроводжує багатьох патологічних станів (еритроцитозу, гіпертромбоцитоз, пошкоджень ендотеліальної судинної стінки різної етіології травматичної, запальної, атеросклеротичної, антифосфоліпідний синдром, пухлин і ін.). Відсутність корекції патологічних станів, які є причиною ГКС, може привести до розвитку гострого тромбозу, інфаркту, інсульту.
27) Порушення тромбоцитарно-судинного гемостазу. Вазопатії. Механізми та клінічні приклади.
При геморагічних вазопатіях уражається судинна стінка, що призводить до порушення тромбоцитарно-судинного гемостазу і кровоточивості, виникає внаслідок підвищення проникності стінки кровоносних судин і її деструкції при порушенні синтезу колагену, при дії біологічно активних речовин, радіотоксінов, імунних геморагічних васкулітах, зниженні ангіотрофічної функції тромбоцитів при тромбоцитопеніях і тромбоцитопатіях, руйнуванні судинної стінки лейкозними інфільтратами. Однією з причин кровоточивості є зменшення вироблення ендотелієм судинної стінки фактора Віллебранда - крупномолекулярний компонента VIII фактора згортання-крові. Цей фактор накопичується в тромбоцитах і звільняється при їх дегрануляції.
Він необхідний для нормальної адгезії тромбоцитів до колагену стінки, і без нього не формується тромбоцитарний тромб. Геморагічний синдром спостерігається і при посиленні перекисного окислення мембранних фосфоліпідів, в результаті чого в ендотелії синтезується і секретується надмірна кількість потужних інгібіторів агрегації тромбоцитів - простациклінів. Крім того, до зниження тромбоцитарно-судинного гемостазу призводить порушення нейрогенної і гуморальної регуляції судинного тонусу, зниження якого веде до неможливості закупорки дрібних судин тромбоцитарним тромбом.
Клінічні прояви – крововиливи у вигляді петехій та пурпур на шкірі та слизових, що виникають унаслідок порушення структур суд. стінки; носові, шлункові кровотечі, мікрогематурія; гіпо- або гіперплазія кісткового мозку з відповідними змінами мегакаріоцитарного ростка; позитивні ендотеліальні проби; збільшений час кровотечі; телеангіектазії.
28) Етіологія та патогенез тромбоцитопеній та тромбоцитопатій. Механізми порушень адгезії, агрегації тромбоцитів, вивільнення тромбоцитарних гранул.
Тромбоцитопенією називається зменшення вмісту тромбоцитів у крові нижче норми (180-320 Г / л). Однак спонтанні кровотечі виникають лише при зниженні їх числа менше 30 Г / л. Під тромбоцитопатіями розуміють якісну неповноцінність і дисфункцію тромбоцитів при нормальному або зниженому їх вмісті.
Етіологія. Причиною виникнення тромбоцитопенії нерідко є імунні реакції при зміні антигенної структури тромбоцитів під дією вірусів, лікарських препаратів, виробленні антитромбоцитарних автоантитіл, несумісності тромбоцитарних антигенів матері і плоду. Крім того, тромбоцитопенія розвивається внаслідок ураження мегакаріоцитарного паростка кісткового мозку іонізуючою радіацією, хімічними речовинами або витіснення його пухлинними метастазами, лейкозними інфільтратами. Зниження тромбоцитопоеза може бути обумовлено дефіцитом ціанокобаламіну і фолієвої кислоти, спадковим дефектом утворення тромбоцитів. Тромбоцитопенія виникає в результаті механічного пошкодження тромбоцитів при спленомегалії, штучних клапанах серця, а також посиленого споживання тромбоцитів при локальному і генералізованому внутрішньосудинному згортанні крові.
До етіологічних чинників, що викликають тромбоцитопатію, відносять дію токсичних речовин і лікарських препаратів, іонізуючої радіації, ендогенних метаболітів; дефіцит ціанокобаламіну, гормональні порушення. Спостерігаються і генетичні дефекти структури мембрани і біохімічного складу тромбоцитів (дефіцит тромбостенина, фактора III, АТФ, АДФ, Г-6-ФДГ, мембранних рецепторів для факторів V, VIII, XI і ін.).
Патогенез. Виділяють чотири основних механізми виникнення тромбоцитопеній: зменшення продукції, посилене руйнування, підвищене споживання, перерозподіл тромбоцитів.
Порушення гемостазу і розвиток кровоточивості при тромбоцитопенії обумовлено наступними механізмами:
підвищенням проникності мікросудин для еритроцитів і інших складових частин крові і ламкість судин внаслідок дистрофії стінки при виключенні ангиотрофическая функції тромбоцитів;
зменшенням адгезивно-агрегаційної функції тромбоцитів;
порушенням реакції звільнення тромбоцитарних факторів згортання крові, АДФ, серотоніну, адреналіну, антігепарінового фактора, наслідком чого є недостатня формування тромбоцитарного тромбу, відсутність спазму судин і уповільнення згортання;
зменшенням ретракції згустку в результаті зниження активності скорочувального білка тромбоцитів - тромбостенина.
У патогенезі ТРОМБОЦИТОПАТІЙ можна виділити два основних механізми їх виникнення - продукція патологічних тромбоцитів в кістковому мозку і деструкція тромбоцитів у всіх відділах системи крові. Патогенез порушення тромбоцитарно-судинного гемостазу при тромбоцитопатіях такий самий, як і при тромбоцитопеніях, так як пов'язаний з виключенням функцій тромбоцитів.
29) Порушення коагуляційного гемостазу: причини та механізми порушень окремих фаз згортання крові. Порушення системи антикоагулянтів та фібринолізу.
Порушення коагуляційного гемостазу, що приводить до розвитку кровоточивості, може бути викликано наступними факторами:
придбаним і спадковим зменшенням або збоченням синтезу плазмових і тромбоцитарних факторів згортання крові та компонентів калікреїн-кінінової системи;
пригніченням або підвищеним споживанням цих факторів;
збільшенням ендогенних антикоагулянтів;
активізацією фібринолітичної системи;
передозуванням антикоагулянтів, фібринолітичних і дефібрінірующего препаратів.
Етіологія. Причинами порушення утворення тромбопластину - є зниження продукції факторів (IX, X) при патології печінки, утворення антитіл до деяких факторів (VIII, IX) при захворюваннях, у патогенезі яких є аутоімунний компонент, або ж передозування такого універсального антикоагулянту, як гепарин. Відомі генетичні дефекти синтезу VIII, IX і XI факторів, дефіцит яких лежить в основі розвитку гемофілії.
Порушення утворення тромбіну - виникає не тільки при захворюваннях печінки, а й при гіпо- та авітамінозах К, коли теж знижується синтез в печінці факторів II, V, VII, що беруть участь в цій фазі. Можлива поява імунних інгібіторів факторів V, VII, посилене їх виведення нирками, спадковий дефіцит (V при Парагемофілія) або ж інактивація компонентами протизгортаючої системи - антитромбін, гепарином (при анафілактичний шок, передозуванні гепарину).
Геморагічний діатез, пов'язаний з порушенням третьої фази освіти фібрину, виникає при зменшенні синтезу фібриногену в уражених патологічним процесом печінки, легенів або ж в результаті спадкової гіпо, афібріногенеміі і дефіциту фібрінстабілізірующего фактора (XIII). Однак значно частіше порушення третьої фази є наслідком посилення фібринолізу при травмі (операції) легких, матки, підшлункової залози; опіку, шоці. Це обумовлено підвищеним надходженням в кров активаторів профібринолізину - тканинних, мікробних фібрінокіназ, лейко- і еритроцитарних активаторів, компонентів калікреїн-кінінової системи і системи комплементу комплексів гепарину з фібриногеном, профібринолізином і адреналіном.
Патогенез. Головними ланками в патогенезі геморагічного діатезу, що розвинувся при порушенні будь-якої з фаз згортання крові, є хронічна крововтрата і її наслідки, а також структурні та функціональні зміни в місці крововиливів.
30) ДВЗ-синдром. Етіологія та патогенез, основні прояви.
Важка патологія гемостазу, що виникає при надмірному надходженні в кров прокоагулянтів і активаторів згортання крові, що веде до утворення множинних мікротромбів в судинах мікроциркуляторного русла, а потім розвитку гіпокоагуляції, тромбоцитопенії і геморагії в результаті "споживання" факторів системи згортання і підвищення функціональної активності системи протизгортання і фібринолізу крові з подальшим виснаженням всіх трьох систем.
Етіологія. Генералізовані інфекції та септичні стани, всі види шоку, травматичні хірургічні операції, акушерська патологія, гострий внутрішньосудинний гемоліз, уремія при нирковій недостатності, все термінальні стану.
Патогенез. Головною ланкою в патогенезі генералізованої гіперкоагуляції є порушення балансу між калікреїн-кініновою, згортання, антизсідальною і фібринолітичною системами крові при надходженні в судинне русло великої кількості прокоагулянтів і їх активаторів. Це і призводить до порушення такої важливої функції крові, як збереження її нормального агрегатного стану, в результаті чого в фазу гіперкоагуляції кров в судинах згортається і припиняється її циркуляція з розвитком важких дистрофічних і функціональних порушень в органах і тканинах, нерідко несумісних з життям. В наступну фазу гіпокоагуляції розрідження крові і втрата здатності до згортання і агрегації тромбоцитів викликають кровотечу, яке погано піддається терапевтичній корекції. За позитивного результату настає третя - відновлювальна фаза, при якій відбувається нормалізація гемостазу.
31) Клінічні форми та етіопатогенез геморагічної хвороби новонароджених.
Геморагічна хвороба новонародженого – це захворювання, що проявляється на 2 – 5-й день життя, обумовлене транзиторним дефіцитом вітамін К-залежних факторів, що супроводжується спонтанними і тривалими кровотечами.
Етіологія:
• знижена активність факторів II, VII, IX і згортання крові (вітамін К- залежні фактори);
• причинами вітамін К-дефіцитного геморагічного синдрому є:
– відсутність вільного вітаміну К у вагітної (при застосуванні лікарських засобів-антагоністів вітаміну К: кумарин, аспірин, фенобарбітал, сульфаніламідні препарати, антикоагулянти);
– порушення всмоктування вітаміну К, при зниженні всмоктування жирів у перші дні життя дитини;
– відсутність бактеріальної кишкової флори, яка синтезує вітамін К;
– пізнє прикладання до грудей;
– тяжкий гестоз у другій половині вагітності;– гепатит у вагітної;
– травматичний розрив пуповини;
– передлежання плаценти або її передчасне відшарування;
– штучне вигодовування.
Патогенез:
• різке зниження рівня протромбіну і факторів II, VII, IX і X згортання крові;
• подовження протромбінового часу, часу згортання крові, часу рекальцифікації плазми;
• залишаються в нормі: час кровотечі, рівень фібриногену, факторів V і VIII, кількість тромбоцитів, ламкість судинної стінки, час рефракції згустка.
Діагностичні критерії:
• кровотечі з шлунково-кишкового тракту;
• носові кровотечі;
• внутрішньочерепні кровотечі;
• кровотечі з пупка;
• блювання кров'ю;
• заочеревинний крововилив;
• пурпура – петехіальні висипання на шкірі та невеликі екхімози (підшкірні геморагії);
• мелена – дьогтеподібне випорожнення, що є результатом кишкової кровотечі;
• крововиливи в легені, печінку, надниркові залози, селезінку;
• кефалогематома.
32.Визначення понять недостатність кровообігу, серцева недостатність. Класифікація. Поняття про гостру та хронічну недостатність кровообігу.
Недостатність кровообігу - це стан, при якому серцево-судинна система не може забезпечити органи й тканини організму необхідною кількістю крові. Є найчастішим проявом різних порушень функцій системи кровообігу.
Недостатність серця - це патологічний стан, обумовлений нездатністю серця забезпечити кровопостачання органів і тканин відповідно до їхніх потреб. Це стан, при якому навантаження на серце перевищує його здатність виконувати роботу.
Класифікація:
I. Залежно від клінічного перебігу розрізняють гостру і хронічну недостатність серця.
II. За вираженістю клінічних проявів недостатність серця може бути прихованою (компенсованою) і явною (декомпенсованою).
III. Залежно від порушення функції переважно того чи того відділу серця розрізняють лівоишуночкову, правошлуночкову і тотальну недостатність серця.
IV. За патогенезом виділяють: а) недостатність серця від перевантаження; б) міокардіальну недостатність серця; в) позаміокардіальну недостатність.
Хворим з гострою серцевою недостатністю потрібна негайна допомога. Хронічна недостатність кровообігу має три ступені:
I ступінь — початкова, прихована, проявляється тільки при значному фізичному навантаженні задишкою, серцебиттям, швидкою втомлюваністю. У стані спокою ці явища зникають.
II ступінь — характеризується у початковому періоді — II (А) — задишкою і серцебиттям при звичайному фізичному навантаженні, набряками ніг, що зникають за ніч, суттєвим зниженням працездатності. Поява задишки у стані спокою, ціанозу, постійних набряків, застійних явищ у черевній порожнині, легенях, розладів функцій органів вказує на перехід захворювання в II (Б) ступінь. Хворі в такому стані непрацездатні.
III ступінь — стан хворого прогресивно погіршується, порушується обмін, відмічається виснаження, відбуваються незворотні зміни у внутрішніх органах. Працездатність повністю втрачена.
33.Види серцевої недостатності (за перебігом, за патогенезом, за переважним ураженням різних відділів серця) – коротка характеристика.
I. Залежно від клінічного перебігу розрізняють:
Гостра недостатність серця розвивається також під час фібриляції шлуночків, пароксизмальної тахікардії, інфаркту міокарда, міокардиту, тромбозу клапанного отвору, емболії легеневої артерії, тампонади серця. При цьому спостерігається недостатнє наповнення кров’ю артеріальної системи, що зумовлює ішемію головного мозку з тяжкими змінами його функції, які нагадують картину шоку і нерідко супроводжуються втратою свідомості та судомами.
Хронічна, або застійна, недостатність серця розвивається поступово, переважно внаслідок метаболічних порушень у міокарді за тривалої гіперфункції серця або різних видів ураження міокарда. Через недостатність викиду крові із серця зменшується кровонаповнення органів на шляхах відтоку. Водночас унаслідок нездатності серця перекачати всю кров, що надходить до нього, розвивається застій на шляхах припливу крові, тобто у венах, скупчується значна кількість крові.
II. Залежно від порушення функції відділу серця розрізняють:
Лівошуночкову розвивається при важких ураженнях м'язу переважно лівого шлуночка або його гемодінамічному перевантаженні. Вона виникає зазвичай як вторинне явище при вроджених вадах серця (коарктація аорти, її стеноз та ін.), набутих вадах мітральних та аортальних клапанів, вторинних артеріальних гіпертензіях, ураженнях міокарда, грубих порушеннях провідної системи серця.
Правошлуночкову є результатом раптового зменшення кровотоку у легенях на фоні різних захворювань та патологічних станів (вроджені вади серця, напади бронхіальної астми, тромбоемболії легеневих артерій, ателектаз легені, стороннє тіло бронхів, гідроторакс та ін.).
Для цього стану характерні блідість, ціаноз, набухання шийних вен, набряки нижніх кінцівок, олігурія. Швидко збільшуються розміри печінки, розширюється права границя серця, з'являються тахікардія, ритм «галопу», зменшуються ударний та хвилинний об'єми серця, знижується АТ.
Тяжкі стани супроводжуються колапсом, ацидозом, порушенням метаболізму, неврологічними розладами, що у сумі знижує резерви серця, скорочувальну здатність міокарда.
IІІ. За патогенезом виділяють:
а) недостатність серця від перевантаження розвивається в результаті дії на здорове серце великих навантажень опором або об'ємом, тобто коли збільшується опір серцевому виштовху або приплив крові до певного відділу серця. Це буває при вадах серця, гіпертензії великого або малого кола кровообігу, артеріовенозних фістулах або при виконанні дуже важкої фізичної роботи. При цьому до серця з нормальною скорочувальною здатністю висуваються надмірні вимоги.
б) міокардіальну недостатність серця розвивається в результаті первинного ураження міокарда. Вона може бути пов'язана з :а)ушкодженням провідникової системи серця (аритмічна) і б)ушкодженням волокон робочого міокарда (міокардіопатична). Причиною її розвитку часто є інфекції, інтоксикація, гіпоксія, авітамінози, порушення вінцевого кровообігу, деякі спадкові дефекти обміну речовин. При цьому недостатність розвивається навіть при нормальному або зниженому навантаженні на серце.
в) позаміокардіальну недостатність розвивається в результаті дії причин, не пов'язаних безпосередньо з міокардом. її виникнення можуть обумовлювати зменшення припливу крові до серця (гіповолемія, колапс) або перешкоди здійсненню діастоли, у результаті чого серце не може прийняти всю кров, що притікає до нього (накопичення ексудату або транссудату в порожнині перикарда, гостра тампонада серця).
34.Вроджені вади серця у дітей, причини їх розвитку – спадкові та набуті в ембріогенезі. Класифікація вроджених вад серця в залежності від характеру порушення гемодинаміки (за Мардером), приклади вад кожного типу.
Вроджені вади серця - це патологічний стан, який характеризується аномаліями розвитку серця та магістральних судин, що виникає внаслідок порушення ембріогенезу в період 2-8-го тижнів вагітності під впливом несприятливих факторів зовнішнього та внутрішнього середовища.
Вроджені — наслідок аномального формування серця і великих судин у перші 3—8 тижнів внутрішньоутробного розвитку плода, якому сприяла шкідлива дія деяких зовнішніх факторів, захворювання матері, природжена спадковість. Деякі вади серця виникають у перші 2—3 місяці життя людини в результаті порушення перебудови механізму кровообігу плоду на самостійний. Основою вроджених вад серця є:
· патологічні сполучення між шлуночками, передсердями і магістральними судинами (дефекти перегородок або їх відсутність, артеріальна протока тощо);
· аномалії клапанів (звуження, облітерація або недостатність);
· звуження або заростання магістральних судин; ненормальне відходження або впадіння великих судин;
· аномалії розташування серця або його камер, аномалії кровопостачання серця.
Набуті вади серця — наслідок захворювань після народження, найчастіше — ревматизму, рідше — атеросклерозу, септичного ендокардиту, сифілісу. Вони виявляються в недостатності клапанів, звуженні (стенозі) клапанних отворів, в комбінації цих вад. Трапляються також дефекти серцевих перегородок, ураження клапанів в результаті поранення або інфаркту міокарда. Найчастіше бувають вади клапана лівого передсердно-шлуночкового отвору, так звані мітральні вади серця, потім — аортальні, рідше — вади інших клапанів.
Класифікація вроджених вад серця (Marder).
Характер порушень гемодинаміки:
ВВС зі збагаченням малого кола кровообігу- дефект міжшлуночкової перетинки(ДМШП),дефект міжпередсердної перетинки(ДМПП),відкрита артеріальна протока(ВАП),
ВВС зі збіднінням малого кола кровообігу- Тетрада Фало
ВВС зі збідненням великого кола кровообігу- коарктація аорти
ВВС без порушення гемодинаміки- аномалія розташування судин
35.Види навантаження на серце. Причини, прояви та можливі компенсаторні реакції.
Виділяють два типи перевантажень серця.
1. Перевантаження об'ємом виникає тоді, коли до серця або до окремих його порожнин притікає збільшений об'єм крові. У цих умовах серце або його відділ, що зазнає перевантаження, мають переміщати збільшений об'єм крові в артеріальну систему. Це досягається збільшенням хвилинного об'єму серця відповідно до збільшеного венозного повернення.
Перевантаження об'ємом виникає при:
а) збільшенні венозного повернення крові до серця, зокрема при збільшенні об'єму циркулюючої крові (гіперволемія) або збільшенні тонусу венозних судин (зменшення ємності венозної системи);
б) вадах серця — недостатності його клапанів. Так, при недостатності аортального і двостулкового клапанів розвивається перевантаження лівого шлуночка, при недостатності клапана легеневої артерії і тристулкового клапана - перевантаження правого шлуночка.
2. Перевантаження опором виникає тоді, коли серце або окремі його відділи змушені виконувати роботу проти збільшеного опору, що перешкоджає переміщенню всієї крові в артеріальну систему. При перевантаженні опором серце має зберегти свій хвилинний об'єм, незважаючи на збільшений опір вигнанню крові.
Перевантаження опором розвивається при:
а) збільшенні артеріального тиску (збільшенні периферичного судинного опору). При гіпертензії великого кола кровообігу перевантаження опором діє на лівий шлуночок, а при гіпертензії малого кола - на правий шлуночок;
б) вадах серця — стенозах клапанних отворів. 'Так, при стенозі отвору аорти розвивається перевантаження лівого шлуночка, при стенозі отвору двостулкового клапана - лівого передсердя, при стенозі отвору легеневої артерії - правого шлуночка, при стенозі отвору тристулкового клапана - правого передсердя.
При дії на серце навантажень об'ємом і опором збільшення роботи серця забезпечується двома типами компенсаторних механізмів.
I. Негайні механізми компенсації серця. До них відносять:
а) гетерюметричтш механізм;
б) гомеометричний механізм;
в) хроноінотропний механізм;.
г) інотропна дія катехоламінів.
II. Механізми довгострокової адаптації серця - гіпертрофія міокарда.
36.Внутрішньосерцеві механізми компенсації підвищеного навантаження та їх характеристика.
Гетерометричпиіі механізм є одним із негайних механізмів компенсації серця до дії навантажень об'ємом. Його сутність полягає в збільшенні сили серцевих скорочень в умовах надходження до серця збільшеного об'єму крові. Основними проявами гетерометричного механізму компенсації є збільшення кінцево-діастолічного тиску за рахунок збільшення надходження крові в порожнини шлуночків і збільшення ударного, а отже, і хвилинного об'ємів серця за рахунок збільшення сили серцевих скорочень. Напруга м'язових волокон міокарда при цьому не міняється. Змінюється тільки їх довжина, звідси назва механізму - гетерометричний.
Гомеометричний механізм є негайним механізмом компенсації серця до дії навантажень опором. Його сутність полягає в збільшенні сили серцевих скорочень в умовах збільшення опору вигнанню крові.
Для гомеометричного механізму характерні такі зміни показників кардіодинаміки:
а) збільшення кінцево-систолічного об'єму;
б) збільшення кінцево-діастолічного об'єму за рахунок первинного підвищення попереднього показника, а не за рахунок збільшення припливу крові, як при гетеро-метричному механізмі;
в) ударний, а отже, і хвилинний об'єми за рахунок збільшення сили серцевих скорочень залишаються на попередньому рівні, незважаючи на збільшення опору вигнанню крові. При гомеометричному механізмі збільшується напруга м'язових волокон міокарда, тимчасом як їхня довжина не міняється. Звідси назва механізму компенсації — гомеометричний.
Хроноінотропний механізм (феномен "сходинок", феномен Боудича) є одним з негайних механізмів компенсації серця до дії підвищених навантажень. Його сутність полягає в тому, що при збільшенні частоти скорочень серця збільшується
сила його скорочень. При цьому одночасно зменшується час розслаблення міокарда, що сприяє швидкому наповненню шлуночків серця кров'ю.
Для хроноінотропного механізму характерні такі зміни показників кардіодинаміки:
а) ударний об'єм збільшується (при навантаженні об'ємом) або залишається постійним (при навантаженні опором). У результаті збільшення частоти серцевих скорочень зростає хвилинний об'єм серця;
б) кінцево-діастолічний об'єм зменшується, якщо приплив крові до серця постійний, або залишається без змін, якщо приплив крові зростає;
в) кінцево-систолічний об'єм зменшується.
37.Позаміокардіальні механізми компенсації підвищеного навантаження та їх характеристики.
Позаміокардіальна недостатність розвивається в результаті дії причин,не пов'язаних безпосередньо з міокардом.Характер.первинноюдіастолічноюдисфункцією . Виникає під дією одного з двох чинників:
1. Зменшення притоку крові до серця по венах. Причини: крововтрата/колапс
2. Неможливість прийняти всю кров, що надходить до серця. Причина- накопичення рідини в порожнинах перикарда.(виділяють швидке/повільне накопичення рідини)
Швидке накопичення зумовлене крововиливом при пораненні/розриві серця, перикардиті, що швидко розвивається. Наслідок: гостра тампонада серця.
Повільне накопичення розв. При хронічному ескудат. перикардиті та гідроперикарді. Наслідок: поступове розтягнення перикарду та збільшення навколосерцевої сумки.
Спостерігається: -зменшення кровонаповнення порожнин серця;
-зниження ударного об*єму;
-падіння АТ(наявна залежність між внутрішньоперикардіальним тиском і АТ: чим більше перший, тим менше другий);
-підвищення венозного тиску.
Компенсаторні механізми:
*При перикардиті відбувається рефлекторно, сигнали надходять з 3 рецепторних полів.
1)отвори порожнистих і легеневих вен- підвищення тиску на шляхах припливу.
2)синокаротидні зони- зниження тиску на шляхах відтоку.
3)перикард- підвищення внутрішньоперикардіального тиску.
*Тампонада серця
Збільшення ЧСС та звуження периферичних судин.
38.Гіпертрофія міокарду, стадії розвитку гіпертрофії за Ф.Меєрсоном
Належить до механізмів довготривалої адаптації серця. Виділяють:
А)Гіпертрофію серця у спортсменів(адаптоване серце). Розв. при періодичних навантаженнях зі зростаючою інтенсивністю. Харкатер. рівномірним збільшенням всіх відділів серця- збалансована. Наслідок: збільшення функціональних резервів серця.
Б) Компенсаторна гіпертрофія серця (переадаптоване серце). Наслідок патологічних процесів, що мають стосунок до серця. Існує два види : гіпетрофія від перенавантаження (при вадах серця, арт. гіпертензії) та від ушкодження( при атеросклеротичних ураженнях, міокардіопатії). Незбалансована. Наслідок: розвиток серцевої недостатності.
Механізм розвитку
Гіперфункція серця→посиленне використання АТФ, що перевищує інтенсивність його ресинтезу→збільшення потенціалу фосфорування →поява/збільшення в клітинах речовин-регуляторів транскрипції→зростання інтенсивності синтезу іРНК та процесів трансляції в рибосомах→посиленний біосинтез структурних, функціонал. білків ті білків-ферментів→збільшення маси міокарду,гіпертрофія.
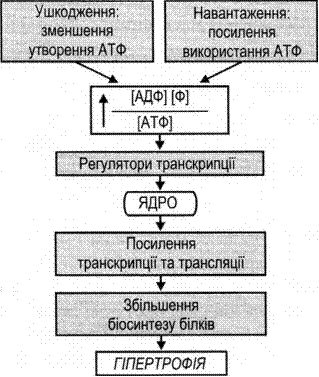
Стадії :
1.Аварійна стадія
Розвивається безпосредньо після підвищення навантаження. Характер. поєднанням патологічних змін в міокарді(зникнення глікогену, зниження рівня креатинфосфату, зменшення вмісту внутрішньокліт. К та підвищення Na, посилення гліколізу, накопичення лактату) з мобілізацією резервів міокарда і організму в цілому. Підвищується навантаження на одиницю м*язової маси→ інтенсивність функціонування структур(ІФС) підвищується. Відбувається швидке, протягом тижня, збільшення маси серця за рахунок посиленого синтезу білків і потвщення м*яз. волокон.
2.Стадія завершеної гіпертрофії і відносно стійкої гіперфункції
Завершено процес гіпертрофії, маса міокарда збільшена на 100-120% і далі не зростає, нормалізується ІФС. Патологічні зміни в обміні речовин і структурі міокарда відсутні, споживання кисню, утвор. енергії, вміст макроергічнихсполук в нормі. Нормалізація гемодинамічних порушень. Серце пристосувалося до нових умов навантаження і протягом тривалого часу компенсує їх.
3. Стадія поступового виснаження та прогресуючого кардіосклерозу
Характеризується глибокими метаболічними і структурними змінами, які накопичуються в енергоутворюючих і скорочувальних елементах клітин міокарда. Частина м*яз.волокон гине та заміщується сполуч.ткан, ІФС зростає. Порушується регуляторний апарат серця. Прогресуюче виснаження компенсаторних резервів зумовлює хронічну серцеву недостатність, далі- недостатність кровообігу.
39.Особливості іннервації, кровопостачання, структури та метаболізму гіпертрофованого серця. Наслідки гіпертрофії.
Гіпертрофоване серце відрізняється низкою обмінних, функціональних і структурних ознак, які дають йому можливість тривалий час долати підвищенне навантаження та створюють передумови для патологічних змін.
1.Відбувається збільшення маси серця за рахунок потовщення м*язових волокон, що супроводжується зміною співвідношення внутрішньоклітинних структур. Збільшення об*єму клітини пропорційно кубу лінійних розмірів, а поверхні- пропорційно їх квадрату призводить до зменшення клітинної поверхні на одиницю маси клітини. Це призводить до погіршення постачання м*яз.волокон, особливо їх центральних відділів.
2.При гіпертрофії знижується інтенсивність росту тубулярної системи і саркоплазматичногоретикулуму, що створює ередумови для порушення процесів скорочення та розслаблення кардіоміоцитів. Внаслідок уповільнення виходу іонів Сф в саркоплазму погіршується скорочення, а через утруднення зворотнього транспорту іонів Са в саркоплазматичнийретикулум- розслаблення.
3.Внаслідок порушення ядерно-цитоплазматичного співвідношення виникають умови для порушення синтезу білків і погіршення пластичного забезпечення клітини.
4. Внаслідок відставання росту маси мітохондрій від росту маси цитоплазми розвиваються деструктивні зміни в міьохондріях, знижується інтенсивність їх роботи, порушується окисне фосфорування. Це призводить до погіршення енергетичного забезпечення гіпертрофованих клітин.
5.Погіршення судинного забезпечення гіпертрофованого міокарду через неадекватне збільшення капілярної мережі відносно збільшеної маси м*язових волокон. Великі вінцеві артеріїї також не здатні забезпечити необхідне пристосування.
6. Порушення регуляторного забезпечення серця зумовлене втягненням в пат. Процес нервового апарату серця. Спостерігається відставання росту нервових закінчень від збідьшення маси скорочувального міокарда. Відбувається виснаження нервових клітин, порушення трофічних впливів, зниження вмісту адреналіну в міокарді, що веде до погіршення його скорочувальних власитвостей, утруднення мобілізації його резервів.
7.Діапазон адаптаційних можливостей гіпертрофованого серця, його здатність пристосовуватися до навантажень, що змінюються обмежені- зменшений функціональний резерв.
Наслідки: виникнення хронічної серцевої недостатності, недостатність кровообігу, атрофія міокарда.
40.Порушення гемо- та кардіодинаміки за умов гострої недостатності кровообігу
До гострої недостатності кровообігу належать гостра серцева недостатність (лівошлуночкова, правошлуночкова, гіподинамічна недостатність при пароксизмальній тахікардії, недостатність кровообігу при глибокій брадикардії, при гіповолемії – після прийому великої дози швидкодіючих діуретиків, при гострій крововтраті), гостра судинна недостатність (шок, колапс, непритомність).
Розрізняють: 1) судинну, 2) серцеву та 3) змішану недостатність.
Серцева недостатність кровообігу – зумовлює зміну багатьох показників гемодинаміки: зменшується ударний і хвилинний об’єм серця внаслідок послаблення скоротливої здатності міокарда;
зменшується надходження венозної крові до серця;
уповільнюється кровообіг;
зменшується ОЦК;
знижується артеріальний систолічний і діастолічний тиск;
змінюється венозний периферичний тиск.
41.Патогенез хронічної серцевої недостатності. Механізми розвитку основних клінічних проявів хронічної недостатності кровообігу(гіпоксії,ацидозу, задишки, ціанозу,набряків).
Внаслідок недостатнього викиду крові з серця зменшується кровонаповнення судин, знижується артеріальний тиск. Водночас через неспроможність серця перепомпувати всю кров, що припливає до нього, розвивається застій в судинах.
Недостатність кровообігу має деякі особливості, взалежності від того, який саме шлуночок уражений – правий чи лівий, і називається відповідно недостатністю за правошлуночковим або лівошлуночковим типом. У першому випадку спостерігається застій крові в венах великого кола кровообігу (збільшується печінка, з’являються набряки на ногах, асцит), у другому – в венах малого кола кровообігу, що може призвести до набряку легень.
Клінічні прояви:
Циркуляторна гіпоксія- кисневе голодування, що розвивається при серцевій недостатності, має характер застійної гіпоксії.
Задишка. Для розвитку симпотому мають значення: а) вплив надлишків іонів Н на хеморецептори судин і безпосередньо на ДЦ; б) набряк інтерстиціальної тканини легень і пов*язане з ним збудження J-рецепторів.
Ціаноз- зумовлений збільшенням концентрації в крові відновленого гемоглобіну в результаті більш повного вилучення кисню тканинами.
Набряки. При правошлуночковій недостатності – розвиваються набряки нижньої половини тіла. При лівошлуночковій- інтерстиціальний набряк легень(серцева астма)/ альвеолярний набряк( синдром набряку легень).
Порушення кислотно-основної рівноваги- можливі всі 4 варіанти змін: а) негазовий ацидоз- внаслідок циркуляторної гіпоксії в крові накопичуються кислі продукти обміну речовин, зокрема , лактату; б)газовий ацидоз- внаслідок альвеолярного набряку легень→ недостатність зовн.дихання→гіперкапнія; в) негазовий алкалоз – зумовлений вторинним гіперальдостеріонізмом і обумовленою ним гіпокаліємією; г) газовий алколоз- настає внаслідок гіпервентиляції, яка призводить до гіпоксії.
42. Недостатність серця від ушкодження міокарду, їх види та прояви.
Недостатність серця — це пат. стан, суть якого полягає в нездатності серця забезпечувати хвилинний об’єм крові, що відповідає потребам органів і ткан.
Міокардіальною назив.недост серця, що розвив в резулт перв ураж серц м’яза.
Прич розв є інфекції, інтоксикація, гіпоксія, авітамінози, порушення вінцевого кровообігу, деякі спадкові дефекти обміну речовин.
У клітинах робочого міокарда відбувається п’ять процесів:
(1) збудження кардіоміоцитів; (2) електромеханічного спряження; (3) власне скорочення; (4) розслаблення; (5) енергозабезпечення міокарда.
1. Порушення збудливості і збудження кардіоміоцитів.
Зміни збудливості волокон міокарда можуть виявляти себе:
1) змінами тривалості потенціалів дії, а отже, і сили серцевих скорочень. Зменш тривалості потенціалів дії виникає при збільш частоти стимуляції, збільше позаклітинної концент іонів калію, при дії ацетилхоліну. Збільш тривалості потенціалу дії характ для охолодження;
1) 2) змінами тривалості періодів абсолютної і відносної рефрактерності, а отже, здатності сприймати імпульсацію (засвоювати ритм). Тривалість цих періодів залежить від тривалості потенціалів дії.
2. Порушення електромеханічного спряження. (Поєднання процесів збудження зі скороченням)
Осн є іони кальцію, що надходять у цитоплазму м’язових клітин під час збудження. У кардіоміо- цитах існує два важливих джерела цих іонів:
а) внутрішньоклітинне — структури сарко- плазматичного ретикулуму: цистерни і поздовжні трубочки (як і в скелетних м’язах). б) позаклітинне — поперечні Т-трубочки і середовище, що оточує м’язові волокна.
Виділяють два механізми електромеханічного спряження в серці.
1. Потенціалзалежне спряження:Суть його полягає в тому, що поширення збудження (потенціалу дії) уздовж плазматичної мембрани зумовлює деполяризацію мембран Т-трубочок, а через них— мембран цистерн і поздовжніх трубочок саркоплаз- матичного ретикулуму. Така деполяризація
цієвих каналів, унаслідок чого іони Са2+ завдяки високому градієнту концентрацій швидко виходять із названих вище структур у цитоплазму.
2. Механізм кальцієвих залпів Під час фази плато потенціалів дії позаклітинні іони Са2+ входять через потенціалзалежні Са-канали в клітину. Цей первинний кальцієвий залп або безпосередньо, або через утворення вторинних посередників (месен- джерів) - 1,4,5-інозитолтрифосфату - діє на мембрани структур саркоплазматичного ретикулуму, викликаючи відкриття наявних там Са-каналів і вихід іонів Са2+ у цитоплазму як наслідок.
На характер електромеханічного спряження в міокарді впливає ряд факторів.
• Тривалість і частота потенціалів дії.
• Концентрація іонів кальцію в позаклітинній рідині.
3. Порушення процесу власне скорочення.( Суть скор кардіоміоцитів полягає в переміщенні актинових і міозинових філаментів одних відносно інших)
Впливають такі чинники:
1) концентрація іонів Са2+ у саркоплазмі
2) ступінь спорідненості тропоніну С до іонів кальцію.
3) стан скорочувальних білків - актину і міозину.
4) концентрація АТФ
4. Порушення процесу розслаблення.(видалення іонів кальцію із саркоплазми, у результаті чого концентрація Са2+ у ній зменшується і стає нижче 10-7 моль/л. При цьому комплекси Са2+ з тропоніном С розпадаються, тропоміозин зміщується по відношенню до актинових філаментів і закриває їхні активні центри - скорочення припиняється.)
Описано три механізми видалення іонів Са2+ із саркоплазми кардіоміоцитів:
1) Са-насоси плазматичної мембрани та саркоплазматичного ретикулуму.
2) Na-Ca-обмінний механізм
3) Са-акумулятивна функція мітохондрій.
Основними причинами порушень розслаблення кардіоміоцитів є:
а) дефіцит АТФ.
б) порушення роботи Са-транспортних систем.
5. Порушення енергозабезпечення. Енергія гідролізу АТФ у функціонально активних кардіо- міоцитах забезпечує:
1) механічну роботу скорочень міофібрил (ковзання міофіламентів один відносно одного);
2) осмотичну роботу - акт транспорт іонів Са2+, Na +, К+ проти градієнтів концентрацій (робота іонних насосів);
3) фосфорилювання білків Са-каналів плазм мембр, фосфоламбану (білка Са- насосів саркоплазматичного ретикулуму).
Розлади енергозабезпечення кардіоміоцитів можуть бути пов’язані з порушеннями:
а) ресинтезу АТФ (гіпоксія, голодування, дефіцит вітамінів, зменшення активності ферментів енергетичного обміну);
б) транспорту АТФ із мітохондрій до місць його використання (порушення креатинкі- назної транспортної системи);
в) утилізації АТФ (зменшення АТФ-азної активності структур кардіоміоцитів).
43. Некоронарогенні ушкодження серця, їх етіологія та патогенез, експериментальне моделювання.
Позаміокардіальною називають недостатність серця, що розвивається в результаті дії причин, не пов’язаних безпосередньо з міокардом.
Вона характеризується первинною діастолічною дисфункцією і може виникати внасл дії одного з двох чинників:
1) зменш притоку крові до серця по венах. Осн прич є гіпово- лемія (крововтрата) або різке розширення судин (колапс);
2) неможл прийняти всю кров, що надходить до серця по венах. (при накопиченні рідини в порожнині перикарда )(запальний ексудат при перикардитах, кров - при пораненнях)
Накоп рід в порожнині перикарда може відбуватися швидко і повільно.
Швидке накопичення відб внасл крововиливу при пораненні або розриві серця, при перикардиті, що швидко розвивається. Через погану розтяжність перикарда в його порожнині підвищується тиск, що перешкоджає діа- столічному розширенню серця, виникає гостра тампонада серця.
Повільне накопичення рідини в перикарді спостерігається при хрон ексуд перикардиті та гідроперикарді, воно супров поступовим розтягуванням перикарда і збільш об’єму навколосерцевої сумки. = внутрішньоперикардіальний тиск не змінюється, а порушення сист кровообігу не виникають тривалий час завдяки більш ефективній у цих умовах роботі компенсаторних мех
44. Патогенез нейрогенних ушкоджень серця.
В основі механізму нейрогенних ушкоджень серця лежить не відповідність між рівнями функції, метаболізму і кровопостачання. Подразнення серцевих симпатичних нервів супроводжується значним збільшенням споживання кисню міокардом. При цьому збільшення вінцевого кровообігу є недостатнім — відносно коронарна недостатність, а тому розвивається гіпоксія міокарда. При склерозування вінцевих артерій невідповідність інтенсивності кровообігу рівневі обміну речовин виявляється ще в більшій мірі, що може виявитися катастрофічним як для серця так і для всього організму в цілому.
45. Коронарогенні ушкодження серця. Недостатність вінцевого кровообігу. Причини та механізми розвитку.
Коронагенні ушкодження серця визначаються значним поширеннням різних форм патології серця і насамперед ішемічної хвороби (ІХС). Коронарна недостатність (КН) - типова форма патології серця, коли коронарні судини не забезпечують потреби міокарда в кисні і субстратах метаболізму.
Недостатність вінцевого кровообігу може бути: а) відносною і б) абсолютною.
-Відносна коронарна недостатність виникає при первинному збільшенні енергетичних потреб серця (збільшення навантаження на серце при фізичній роботі, артеріальній гіпертензії). При цьому інтенсивність вінцевого кровотоку зростає, але є недостатньою для задоволення зрослих потреб серця.
-Абсолютна коронарна недостатність виникає при первинному порушенні вінцевого кровообігу, у результаті чого зменшується доставка кисню і поживних речовин до міокард як у стані спокою, так і при збільшенні енергетичних потреб серця.
П р и ч и н н і фактори КН можна розділити на 2-і групи:
1) Коронарогенні.
2) Некоронарогенні.
• Із коронарогенними факторами пов‘язане зменшення або повне закриття просвіту вінцевих артерій і в зв’язку з цим - значне зниження припливу артеріальної крові до міокарда. Такі фактори обумовлюють розвиток абсолютної КН. До них належать:
1) Атеросклеротичні ушкодження стінок коронарних артерій
2) Агрегація формених елементів крові (головним чином еритроцитів і тромбоцитів) і утворення тромбів у вінцевих артеріях серця.
3) Спазм коронарних артерій.
4) Зниження перфузіїного тиску в коронарних артеріях
• Некоронарогенні фактори викликають істотне підвищення «потреб» і витрат міокардом кисню та субстратів метаболізму в порівнянні із рівнем їх надходження. Вони обумовлюють розвиток відносної КН.
1) Підвищенням у крові і міокарді рівня катехоламінів
2) Значним зростанням роботи серця
Механізми (патогенез) ушкодження міокарда при коронарній недостатності.
Значне збільшення витрати кисню і субстратів обміну речовин міокардом, а також порушення відтоку продуктів порушеного обміну речовин, іонів, біологічно активних сполук в умовах КН обумовлює розвиток гіпоксії, що внеде до «включення» ряду загальних механізмів ушкодження міокарда: 1) розлад процесів енергетичного забезпечення кардіоміоцитів; 2) ушкодження їх мембранного апарату; 3) альтерацію ферментних систем клітин; 4) дисбаланс іонів і рідини; 5) розлад механізмів регуляції роботи серця.
Отже, патогенетичною основою КН - є ішемія і гіпоксія міокарда.
46. Ішемічна хвороба серця, її прояви. Стенокардія, інфаркт міокарду.
Ішемічна хвороба серця — це хвороба, що розвивається в результаті абсолютної недостатності вінцевого кровообігу й виявляється ушкодженнями міокарда різного ступеня тяжкості.
ЇЇ основними клінічними проявами є:
1) стенокардія — напади короткочасної (до 20 хв.) гострої коронарної недостатності, які супроводжуються больовим синдромом, відчуттям страху і пов'язаними з цим вегетативними реакціями.
Розрізняють стенокардію напруги, стенокардію спокою і стенокардію Принцметала (спазм вінцевих артерій);
2) передінфарктний стан (проміжний коронарний синдром, або гостра вогнищева дистрофія міокарда) — розвивається при тривалості'ішемії міокарда від 20 до 40 хв.;
3) інфаркт міокарда— некроз серцевого м'яза, обумовлений порушеннями вінцевого кровообігу. Виникає при оборотній (транзиторній) ішемії, що триває понад 40-60 хв. або при необоротних порушеннях коронарного кровообігу;
4) кардіосклероз — склеротичні зміни серцевого м'яза. Можуть бути дифузними (атеросклеротичний кардіосклероз) і вогнищевими (постінфарктний кардіосклероз).
СТЕНОКАРДІЯ
Стенокардію характеризують:
-пароксизмальність;
-короткочасність;
-чіткий і швидкий терапевтичний ефект від прийому нітратів.
ЕТІОЛОГІЯ
• атеросклероз вінцевих артерій
• вазоспазм
• порушення в системі гемостазу (зміна функції тромбоцитів, підвищення згортання активності крові, пригнічення фібринолізу)
• недостатньо розвинена мережа колатерального кровообігу
• гіперпродукція катехоламінів.
Фактори ризику:
Спадкова обтяженість по артеріальній гіпертонії, ІХС
Дисліпідемії (гіперхолестеринемія)
Куріння, Ожиріння , Гіподинамія , Цукровий діабет
В основі ПАТОГЕНЕЗУ лежить невідповідність між потребою міокарда в харчуванні і здатністю коронарних судин забезпечити адекватний кровотік.
КЛІНІЧНІ ПРОЯВИ СТЕНОКАРДІЇ
◄ Нападоподібний характер болю (раптово починається і поступово вщухає) ◄ Локалізація болю за грудиною
◄ Короткочасність нападу - 1-10 хв. Затяжний больовий напад вимагає виключення іншої форми ІХС - інфаркту міокарда.
◄ Припинення нападу після припинення навантаження або через 1-3 хв. після сублінгвального прийому нітрогліцерину
Стенокардію класифікують на:
◄ Стенокардія напруги
◄ Вперше виникла стенокардія (больовий синдром тривалістю менше 1 місяця.)
◄ Прогресуюча стенокардія напруги (збільшення частоти і тривалості нападів при однаковому ступені фізичного навантаження.
◄ Спонтанна стенокардія (варіантна стенокардія, напади виникають в нічні години)
◄ Рання постінфарктна стенокардія (від 3 до 28 діб).
КЛІНІЧНІ ФОРМИ СТЕНОКАРДІЇ
I Ф.К. стенокардії напруги: повсякденна навантаження не викликає нападів захворювання. Біль за грудиною виникає при надмірних або тривалих фізичних навантаженнях (при прискореної ходьбі або ходьбі середнім кроком до 1000 м (ВЕМ - 750 кгм / хв.)
II ф.к.: невелике обмеження повсякденної діяльності. Напад стенокардії може виникнути при швидкій ходьбі, підйомі по сходах, особливо після прийому їжі (при ходьбі по рівній місцевості до 500 м (ВЕМ - 450 кгм / хв) Ознаки захворювання можуть також з'явитися в стані стресу, в холодну погоду або в перші кілька годин після пробудження.
III ф.к. : значне обмеження фізичної активності. Підйом через один проліт сходів або нетривала ходьба по рівній місцевості викликають появу болю (при ходьбі по рівній місцевості до 150-200 м (ВЕМ - 150-300 кгм / хв).), Задишки і змушують хворого зробити перепочинок. ◄ IV ф.к.: практично повне обмеження повсякденної активності. Практично будь-яка, навіть найлегше фізичне навантаження викликає приступ «грудної жаби» у хворого.
ІНФАРКТ МІОКАРДА
За глибиною і поширеністю некрозу розрізняють:
великоосередковий (з патологічним зубцем Q, трансмуральний)
дрібноосередковий І.м. (без патологічного зубця Q, субендокардіальний, інтрамуральний).
За локалізацією ІМ буває передній, верхівковий, бічний, септальний, задньої стінки (діафрагмальної ділянки) і задньобазальний. Можуть бути також поєднані ураження. Залежно Залежно від характеру перебігу розрізняють ІМ із затяжним розвитком, рецидивуючий і повторний.
При І.м. виділяють такі періоди:
-найгостріший (перші ознаки некрозу виникають від 30 хв до 6 год після появи ішемізованої ділянки міокарда);
-гострий (перші 10 днів від початку захворювання);
-підгострий період (з 10-го дня до кінця 8-го тижня, коли завершуються початкові процеси формування рубця);
-постінфарктний період (характеризується збільшенням щільності рубця та адаптацією серця і продовжується від 2–6 міс з моменту утворення рубця до 2–3 років).
Причини:
1. Атеросклероз вінцевих артерій . Його розвиток супроводжується порушенням постачання міокарда киснем.
2. Збільшення навантаоїсення на серце (фізична напруга, артеріальна гіпертензія). При цьому збільшується потреба серця в кисні.
3. Стрес
Клінічні синдроми:
1. Больовий синдром.
2. Гостра серцева недостатність. Розвивається при уражені великих ділянок міокарда. Може виявляти себе синдромом серцевої астми і набряку легень або карді-огенним шоком.
3. Аритмічний синдром. Можливий розвиток усіх видів аритмій. Найнебезпечнішою є поява фібриляції шлуночків.
4. Резорбційно-н
екротичний синдром.
КЛАСИФІКАЦІЯ
Згідно з МКХ-10 серед різновидів гострого ІМ виділяють:
- гострий ІМ з наявністю патологічного зубця Q (I21.0-I21.3);
- гострий ІМ без патологічного зубця Q (I21.4);
- гострий ІМ (неуточнений — у разі утрудненої діагностики I21.9);
- рецидивний ІМ (
- повторний ІМ (I22);
- гостра коронарна недостатність (проміжний I24.8).
Патогенез гострого ІМ.
Розвитку коронаротромбозу передує пошкодження ендотелію атеросклеротичної бляшки з підвищенням в крові тромбогенних факторів (тромбоксану А2 тощо). Сприяють пошкодженню гемодинамічні порушення, різні зміни тонусу вінцевих судин, пов'язані з діяльністю серця, і коливання рівню катехоламінів. Розрив атеросклеротичної бляшки з некрозом в центрі і пристінковим тромбозом, помірно вираженим стенозуванням є досить прогностичне небезпечним, нерідко закінчується або ІМ, або раптовою коронарною смертю. В першу чергу до розриву схильні новоутворені атеросклеротичні бляшки, багаті на атерогенні ліпопротеїди. Частіше ІМ діагностують у ранкові години, що пов'язано з циркадними змінами тонусу коронарних судин і концентрації катехоламінів.
Згідно з динамічними змінами на ЕКГ, розрізняють такі стадії гострого ІМ :
І. Стадія пошкодження (найгостріша) — від початку підйому сегмента ST до формування патологічного зубця Q.
II. Гостра стадія — наявність зубця Q, підйом ST і злиття його з зубцем Т.
III. Підгостра стадія — наявність зубця Q, повернення ST на ізолінію, негативний, «коронарний» Т.
IV. Рубцева стадія — наявність зубця Q з позитивним зубцем Т.
47.Прояви та ускладнення інфаркту міокарда. Некротично-резорбційний синдром, синдром Дресслера.
Ускладнення інфаркту.
Ранні
1)Порушення ритму і провідності( спостерігаються екстрасистолія, пароксизмальна тахікардія, миготлива аритмія, порушення провідності, аж до повної поперечної блокади)
2)Кардіогенний шок( гостра недостатність кровообігу, викликана різким зниженням скоротливої здатності міокарда внаслідок первинного ураження серця, бо порушенням ритму його діяльності)
3)Гостра серцева недостатність
4)Розрив серця
Піздні
1)Синдром Дресслера( розвиток перикардиту, плевриту і пневмоніту. Причиною стає накопичення в крові антикардіальних аутоантитіл. В крові лейкоцитоз, еозинофілія, підвищенням ШОЕ)
2)Хронічна недостатність кровообігу
3)Аневризма серця
4)Тромбендокардіт і тромбоемболії
Серцевий напад(інфаркт)- пекучий чи стискаючий біль або як дискомфорт і важкість за грудиною. Біль триває кілька хвилин може знову з’явитися.
48.Можливі наслідки ішемії міокарду. Реперфузійний синдром, етіологія та патогенез.
1)Розвиток серцевої недостатності
2)Електрична нестабільність серця(розвиток аритмій)
3)Ушкодження кардіоміоцитів
4)Реперфузійний синдром(виникає внаслідок поновлення кровообігу вішемізованих ділянках, обумовлене припиненням коронарного ангіоспазму, мінімальна тривалість ішемії після якої виникає реперфузійний синдром-40хв. Якщо ішемія 20-40 хв-реперфузійні ушкодження серця.Основою є так званий кисневий парадокс.(різка активація процесів пероксидного окиснення ліпідів(надходження кисню в клітини), скидання електронів на молекули кисню=вільні радикали=ініціюють реакції пероксидного окиснення ліпідів(ушкодженняя клітинних мембран))
49.Гостра серцева недостатність, етіологія, основні прояви.
Вроджені та набуті вади серця, фіброеластоз міокарда, міокардити, перикардити, обмінні кардіопатії, міокардіодистрофія,захворювання легенів, печінки, нирок, органів кровотворення та інші хвороби, важка травма головного мозку,порушення цілісності клапанів або камер серця; тампонада серця.
1)у великому колі кровообігу -периферичні набряки (тістоподібні набряки навколо кісточок або крижової ділянки; можуть не встигнути розвинутись), розширення яремних вен, пальпаторна болючість в епігастрії (внаслідок збільшення печінки)
2) у малому колі кровообігу - задишка, прискорене, хрипи над легеневими полями3) зниженого серцевого викиду- швидка стомлюваність, відчуття слабкості, аменція, сонливість,шкіра бліда, гіпотензія, олігурія
50.Порушення ритму серця. Види аритмій.
Зміни нормальної частоти, регулярності та джерела збудження серця, порушення проведення імпульсів, зв’язку або послідовності між активацією передсердь і шлуночків- порушення ритму серця. Нормальний автоматизм=синусним вузлом провідної системи- атріовентрикулярний вузол-пучок Гіса і його відгалуження- волокна Пуркіньє
Аритмії:
1)Дихальна аритмія (Збільшення ритму пульсу під час вдиху повітря та його уповільнення на видиху - ось ознаки такої аритмії. Проявляється у людей середнього віку і в кого підвищена збудливість вегетативної нервової системи)
2)Миготлива аритмія (це захворювання, при якому відбувається швидке та нерегулярне скорочення передсердь =скорочувальна функція серця погіршується. Миготлива аритмія може проявлятись у вигляді окремих нападів або ж спостерігатись постійно, інколи навіть впродовж багатьох років. Інколи першим проявом миготливої аритмії може бути інсульт)
3)Синусова тахікардія(виникає після фізичних навантажень, емоційних перенапружень, підвищеній температурі при застудних захворюваннях. У людини серцевий м’яз може скорочуватись понад 90 ударів на хвилину.)
4)Синусова брадикардія (вид аритмії, протилежний синусовій тахікардії. Серцевий м’яз скорочується до 55 ударів на хвилину. Проявляється при захворюваннях щитовидної залози. Загальна слабкість та запаморочення – головні ознаки синусової брадикардії.)
5)Синусова аритмія(Найчастіше проявляється у дітей та підлітків. Вона не потребує лікування, а діагностується затримкою дихання, під час якого зникає.)
51.Особливості проявів, причини розвитку аритмій у дітей. (Причини, з яких найчастіше зустрічаються у дітей аритмії).
Аритмія в дітей може виникають в будь якому віці, найбільший ризик з розвитку та проявів аритмії є період відразу після народження і від 4 до 5 років, від 7 до 8 років і з 12 до 14 років. Саме тому в рамках диспансеризації дітей цього віку доцільно проводити обов'язкові консультації дитячого кардіолога та ЕКГ-скринінг.Можливі вроджені вади серця (аномалія Ебштейна, дефект міжпередсердної перегородки, відкритий атріовентрикулярний канал, тетраду Фалло). Виникнення аритмій може розвиватися внаслідок міокардитів, міокардіодистрофії, дилятаційної та гіпертрофічної кардіоміопатії, перенесеного ревматизму, через пухлини серця, перикардити, травми серця, що супроводжуються крововиливом в область провідних шляхів, інтоксикацією, ангіна, дифтерія, пневмонія, бронхіт, кишкові інфекції, сепсис, що супроводжуються втратою рідини та призводять до електролітних порушень. Механічні дії при зондуванні порожнин серця та проведенні ангіографії. У дітей можливі вроджені порушення ритму, обумовлені аномаліями розвитку провідної системи, аритмогенною дисплазією правого шлуночка. Екстракардіальнимифактори - патологічний перебіг вагітності та пологів, недоношеність, внутрішньоутробна гіпотрофія плода,функціональні розлади нервової системи, захворювання крові.Змішаного типу аритмія-поєднання органічних захворювань серця та порушень нейрогуморальної регуляції його діяльності.Синусова аритмія в дитини може бутиреакцією організму на спекотну погоду, неадекватне фізичне навантаження, сильні емоції.
52. Синдром подовженого інтервалу QT. ЕКГ прояви, ускладнення
(поліморфна шлуночкова тахікардія, або “піруетна тахікардія”),
етіологія, можливі клінічні наслідки, лікування.
LQTS (long Q-T syndrome – LQTS) являє собою подовження інтервалу
Q-T ЕКГ. Синдром подовженого інтервалу QT найчастіше є наслідком
генетично запрограмованих порушень у калієвих або натрієвих іонних
каналах і в типових випадках маніфестується подовженням інтервалу QT
на стандартній поверхневій ЕКГ.
Тривалість інтервалу Q-T на ЕКГ – електрична систола серця – сумарно
відображає процеси деполяризації і реполяризації в кардіоміоцитах, що
виникають у результаті руху електролітів у клітину ззовні і навпаки, який
контролюється К+-, Na+-, Ca2+-каналами сарколеми, енергетичне
забезпечення яких здійснюється Mg2+-залежною АТФазою. Причини
порушення цих процесів ведуть до подовження інтервалу Q-T –
сповільненої і асинхронної реполяризації міокарда шлуночків. На цей час
численні дослідження підтверджують, що вроджений LQTS є генетично
гетерогенним захворюванням. У літературі описано близько 180 мутацій,
що локалізуються в 6 генах, розташованих переважно на трьох
хромосомах: 7, 11 і 3.
Для діагностики вроджених форм LQTS у випадку граничного подовження
інтервалу Q-T і(або) відсутності симптомів Р. Schwartz в 1985 р.
запропонував набір діагностичних критеріїв [20]. “Великі” критерії – це
подовження коригованого інтервалу Q-T (Q-Tс) понад 440 мс, наявність в
анамнезі епізодів синкопе і синдрому подовженого інтервалу Q-T у членів
сім’ї. “Малі” критерії – це вроджена нейросенсорна глухота, епізоди
альтернації зубця Т, повільний серцевий ритм (у дітей) і патологічна
шлуночкова реполяризація. У 1993 р. ці критерії були переглянуті з
урахуванням залежності тривалостіінтервалу Q-Tс від статі пацієнтів.
Значення Q-Tc понад 440 мс для чоловіків і понад 460 мс для жінок
вважають збільшеними [28]. Найбільше діагностичне значення мають
достовірне подовження інтервалу Q-T, пароксизми шлуночкової тахікардії
“torsade de pointes” і епізоди синкопе.
Ускладнення: пароксизми шлуночкової тахікардії типу “пірует” (“torsade
de pointes” - наростання і зменшення амплітуди комплексів QRS), що
клінічно проявляються синкопальними станами.
P.S. Синкопальні стани – це синдром, який характеризується
короткочасною і раптовою втратою свідомості, зазвичай
супроводжується втратою м’язового тонусу і падінням.
Поліморфна тахікардія - комплекси QRS під час тахікардії змінюють свій
вигляд від скорочення до скорочення.
поліморфна шлуночкова тахікардія
Етіологія:
1. Вроджений синдром подовженого інтервалу Q-T:
а) генетичні форми: синдроми Романо–Уорда
(аутосомно-домінантний тип) і Джервела–Ланге–Нільсена
(поєднується з вродженою глухонімотою);
б) спорадичні форми.
2. Набуті форми синдрому, що виникають внаслідок:
● а) прийому лікарських препаратів:
○ антиаритмічних (хінідин, дизопірамід, енкаїнід, флекаїніду
ацетат, бепридил, етацизин),
○ фенотіазинів,
○ трициклічних антидепресантів,
○ препаратів літію;
● б) порушення метаболізму;
в) низькокалорійної дієти;
г) захворювань центральної і вегетативної нервової системи;
д) захворювань серцево-судинної системи:
○ ішемічної хвороби серця,
○ пролапса мітрального клапана.
Вважають, що набутий подовжений інтервал Q-T утричі частіше
спостерігається у чоловіків і більш характерний для осіб похилого і
старечого віку із захворюваннями, при яких домінуючими є коронарогенні
ураження міокарда.Останнім часом з’явилися праці, в яких виникнення
набутого LQTS пов’язане з присутністю у хворих “мовчазної” мутації в
одному з генів, що відповідає за вроджений LQTS. Зареєстровані випадки,
коли прийом медикаментів призводив до маніфестації безсимптомного
раніше вродженого LQTS.
Гіпотеза симпатичного дисбалансу – порушується правостороння
симпатична іннервація серця. В результаті формується асиметрія
іннервації з перевагою лівосторонніх симпатичних впливів. Зв’язок цього
порушення з подовженням інтервалу Q-T на ЕКГ був переконливо
доведений в експериментах з видаленням правосторонніх симпатичних
гангліїв.
Первинний синдром LQTS – це спадкова патологія. У сім’ях з такою
спадковістю відзначається висока концентрація випадків раптової смерті у
молодому віці, повторні випадки втрати свідомості у родичів різного
ступеня спорідненості. При цьому раптова смерть у дітей може виникати в
момент активного заняття спортом, плавання, емоційного збудження або у
них часто реєструють втрату свідомості з дня народження. По-різному
захворювання проявляється не тільки у людей різного віку, а й різної статі.
У хлопчиків втрата свідомості буває частішою, важчою і небезпечнішою.
Багато з них при відсутності лікування не доживають до 16–18 років, до
репродуктивного періоду. З огляду на це, поширилася думка про те, що
захворювання передається виключно по материнській лінії.
53. Порушення автоматизму. Класифікація, патогенез, прояви.
Порушення автоматизму серця - це патологія провідникової системи
серця.
1. Номотопні аритмії: генерація імпульсів до скорочення, як і в нормі,
відбувається в синусно-передсердному вузлі. До цієї групи
відносять:
а) синусну тахікардію - збільшення ЧСС
б) синусну брадикардію - зменшення ЧСС
в) синусну (дихальну) аритмію - зміну ЧСС у різні фази дихального
циклу (прискорення при вдиху і уповільнення при видиху).
2. Гетеротропні аритмії - синдром слабкості синусно- передсердного
вузла.
Генерація імпульсів до скорочення відбувається не в
синусно-передсердному вузлі, а в інших структурах провідникової
системи, що є водіями ритму ІІ і ІІІ порядку. Синдром розвивається
в результаті зменшення активності або припинення діяльності
синусно-передсердного вузла при ушкодженні його клітин або
первинних функціональних порушеннях. При цьому можуть
розвиватися такі види патологічних ритмів серця:
а) передсердний повільний ритм - водій ритму міститься в
структурах лівого передсердя, ЧСС < 70 уд/хв.;
б) атріовентрикулярний ритм - джерелом імпульсів є водії ритму ІІ
порядку (верхня, середня або нижня частина АВВ), ЧСС залежно від
місця генерації імпульсів зменшується від 70 до 40/хв.
в) ідіовентрикулярний шлуночковий ритм - генерація імпульсів
відбувається у водіях ритму ІІІ порядку (пучок Гібса або його
ніжки), ЧСС < 40/хв.
Зміна показників, від яких залежить ЧСС (максимальний діастолічний
потенціал, критичний потенціал та швидкість діастолічної деполяризації)
веде до зміни частоти генерації імпульсів або до появи інших джерел і
пульсації, якщо ці зміни виникають в інших, здатних до збудження
ділянках серця і призводить до появи в них потенціалів дії. При зменшенні
рівня максимального діастолічного потенціалу клітин САВ. При
наближенні до нього максимального діастолічного потенціалу або
54. Екстрасистоли: механізм виникнення ектопічних вогнищ
збудження, види екстрасистол та основні особливості ЕКГ при різних
видах екстрасистол.
Екстрасистолія - це вид аритмій, обумовлених порушенням функції
збудливості, що виявляє себе виникненням позачергових скорочень серця
або тільки шлуночків.
Екстрасистола – вид аритмій, зумовлених порушенням провідності, що
проявляється виникненням позачергових скорочень серця, чи тільки
шлуночків.
І Синусна. Внасл передчасного збудження частини кл сино-атріального
вузла. ЕКГ – не відрізн від норм, зменш діаст інтервалу Т-Р. Вкороч
діастола, зменш наповнення шлуночків – зменш пульсова хвиля.
ІІ Передсердна. Наявність вогнища ектопічного збудження в різних
ділянках передсердь. ЕКГ: викривлення ф-ми Р, збережений QRST,
подовж діаст інтервал після екстрасист. Збудження йде ретроградно,
передчасно розряджає норм синус імпульс, що збігається зі збудж шлун. Є
неповна компенс пауза.
ІІІ Передс-шлуночкова. Виникнення додатк імп в передс-шлун вузлі.
Хвиля збудж поширюється у шлун – нормально, у передс – ретроградно.
Негативний Р, може збігатись з QRST. Діастол інтервал подовжений.
IV Шлуночкова. Є повна компенс пауза – внасл того, що збудж, що
охоплює шлун, не передається через передс-шлун вузол на передс,
черговий норм імпульс з сино-атр вузла не поширюється на шлуночки, які
– в фазі рефрактерності.
55. Порушення провідності. Блокади та основні електрокардіографічні
прояви атріовентрикулярних блокад І, ІІ, ІІІ ступеню. Повна
атріовентрикулярна блокада, синдром Морганьї-Адамса-Стокса.
Блокади – уповільнення або повне припинення проведення імпульсів по
якій-небудь ділянці провідної системи серця. Етіологія виникнення
блокадрізноманітні захворювання серцево-судинної системи: на першому
місці по частоті порушень функції провідності коштують різні форми ІХС,
далєє йдуть запальні кардіоміопатії з міокардитичним кардіосклерозом, а
також метаболічні і морфологічні кардіоміопатії і ін. захворювання. По
локалізації виділяють: - блокади синоатриальні (-синоаурикулярні) -
внутрішньопередсердні (точніше міжпередсердні, оскільки в даному
випадку порушується проведення імпульсу з правого на ліве передсердя
по лівій гілочці пучка Бахмана) - атріовентрикулярні -
внутрішньошлуночкові. 37 Залежно від тяжкості порушень проведення
імпульсу блокади можуть бути I, II, III ступені.. Неповна блокада I ступеня
– збільшується час проведення імпульсу через «блокадну» ділянку, але всі
імпульси проходять. Неповна блокада II ступеня – частина імпульсів
випадає. Залежно від закономірності випадання імпульсів вузлові блокади
(синоатріальні і атріовентрикулярні) II ступені, які підрозділяються
залежно від того, має місце на ЕКГ періоди Венкебаха – Самойлова або
вони відсутні на два типа-MOBIZ І і MOBIZ ІІ . Внутрішньошлуночкові
блокади діляться на повні і неповні, а також виділяють скороминущі
(транзиторні), переміжні (інтермітуючі) і латентні блокади . Блокади III
ступеня (повні) - повне припинення проведення імпульсів в ділянці
провідної системи. Клінічна класифікація порушень провідності серця:
ПОРУШЕННЯ ПРОВЕДЕННЯ ІМПУЛЬСУ Синоаурікулярні блокади - І
ст. і ІІІ ст., які по загальноприйнятій ЕКГ не виділяються. - ІІ ст. першого
типу (Mobiz І) і другого типу ( Mobiz ІІ). Внутрішньопередсердні блокади
- неповні - повні Атріовентрикулярні блокади: I ст. II ст. - першого типу
(Mobiz 1), другого типу (Mobiz 2) субтотальні (майже повні),коли
неможливо визначити за яким типом відбувається випадіння ( який Mobiz?
). ІІІ ст. - повна поперечна блокада Внутришлуночковые блокади:
Однопучкові (монофасцікулярні): блокада правої ніжки пучка Гіса
блокада передньої гілки лівої ніжки пучка Гіса блокада задньої гілки лівої
ніжки пучка Гіса - посттійна -минуча 38 двупучкові ( біфасцикулярні):
одностороння: блокада лівої ніжки пучка Гиса двосторонні: блокада
правої ніжки пучка Гіса і передньої гілки лівої ніжки пучка Гіса (блокада
Вільсона). блокада правої ніжки пучка Гіса і задній гілці лівої ніжки пучка
Гіса трьохпучкові (трифасцікулярні) неповна. повна. Синоатріальна
блокада (сино-аурикулярна) – порушення проведення імпульсу з
синусового вузла на передсердя. Основні її етіологічні чинники : ІХС,
міокардит, миокардитичний кардіосклероз, інтоксикація серцевими
глікозидами, роздратування блукаючого нерва. Клінічна картина залежить
від основного захворювання і ступеня блокади. При II, III ступені під час
пауз в серцевій діяльності можуть розвиватися напади
Морганьї-Адамс-Стокса, що вимагають невідкладних заходів. Інші прояви
С-А-блокади – серцебиття, перебої в роботі серця, брадикардія,
порушення гемодинаміки. I ступінь С-А-блокади по загальноприйнятій (12
відведень) ЕКГ, не виявляється, діагностика можлива тільки при
спеціальному елекрофізіологічному обстеженні. II ступінь С-А-блокади.
Розрізняють два ЕКГ – варіанти: С-А-блокади першого типу або MOBIZ І і
другого типу або MOBIZ ІІ. До першого типу відносить С-А-блокади з
періодами Венкебаха-Самойлова, до II типу – без періодики. Періодика
Венкебаха-Самойлова це властивість провідної системи, яка
характеризується прогресуючим погіршенням проведення імпульсу по
ділянці провідної системи від імпульсу до імпульсу,перед його
припиненням або блокуванням. При сино-атріальних блокадах окремі
імпульси не виходить з синусового вузла і не викликає збудження ні
передсердя, ні шлуночків. 39 На ЕКГ С-А-блокада II ступеня ( Mobiz ІІ)
без періодики ВенкебахаСамойлова виявляється періодичним випадінням
одного (або декілька) серцевих циклів P-QRS-Т. Виникаюча пауза
(інтервал Р-Р(або R-R) під час асистолії) рівна подвоєному (потрійному і
так далі) основному інтервалу Р-Р(R-R). Часто під час пауз реєструються
вислизаючі (що заміщають) комплекси або ритми. Періоди
Венкебаха-Самойлова при С-А-блокаді II ступеня (Mobiz І) виявляються
прогресуючим скороченням інтервалу Р-Р(R-R) перед випаданням всього
серцевого цикла- P-QRS-T. Пауза менше подвоєного найкоротшого
інтервалу Р-Р перед випаданням. Скорочення Р-Р перед паузою
характерний для типової сино-атріальної блокади ІІ ступені і засновано на
математичній закономірності .Суть математичної закономірності в тому,
що хоча час проведення імпульсу по «блокадній» ділянці з кожним
імпульсом зростає, проте «приріст» за часом (інкремент) від імпульсу до
імпульсу зменшується. У зв’язку з тим, що при цій блокаді відсутній той
інтервал по котрому можна судити про прогресуюче погіршення
провідності узята аналогія з A-V блокадами. По цьому інтервал Р-Р перед
паузою буде найкоротший, а інтервал Р-Р після паузи щонайдовший.
Спостерігаються також атипові періодики, серед яких частіше
зустрічається варіант з подовженням останнього Р-Р перед випаданням
P-QRS-T. III ступінь С-А-блокади виявляється на ЕКГ заміщуючим
ритмом з центрів автоматизму(тому, по загальноприйнятій ЕКГ цей
ступінь блокади також не виділяється, діагностується. інший ритм, не
синусовий), другого порядку( найчастіше передсердні ритми). Тому СА
блокади III ступінь теж не виділяються, за винятком того випадку коли
при моніторному ЕКГ спостереженні фіксується момент виникнення
повної сино-атріальної блокади, спостерігається на ЕКГ ізолінія і потім
бере на себе функцію водія ритму центр автоматизму, що пролягає нижче
56. Одночасне порушення автоматизму та провідності. Тремтіння,
миготіння передсердь. Миготлива аритмія. Фібриляція шлуночків.
I. Аритмії, обумовлені порушенням автоматизма
1. Синусова тахікардія. 2. Синусова брадикардія 3. Синусова аритмія.
II.Ектопічні ритми: 1. Предсердна екстрасистолія. 2. Атриовентрикулярна
екстрасистолія. 3. Шлуночкова екстрасистолія.
III. Пароксизмальна і непароксизмальна
1. Передсердна форма. 2. З атриовентрикулярного з‘днання. 3.
Шлуночкова форма.
IV. Фібриляція і тріпотіння:
1. Фібриляція передсердя. 2. Тріпотіння передсердя. 3. Тріпотіння і
фібриляція шлуночків.
V. Порушення функциї провідності:
1. Атриовентрикулярна блокада. 2. Блокади ніжок пучка Гіса.
VI. Синдроми передчасного збуждення шлуночків.
Фібриляція передсердя (миготлива аритмія) 68 Основні причини: ІХС,
мітральний стеноз, тіреотоксикоз ( у сумі – в 98% випадків), а також при
ділятаційній кардіоміопатиї, алкогольної кардіоміопатії та інш. При
даному порушенні ритму відсутні скорочення передсердя як єдиного
цілого, а має місце збудження і скорочення окремих груп м'язових волокон
(систола передсердь відсутня).. Частота їх хаотичних скорочень складає
від 350 до 700 в одну хвилину. Різні волокна передсердя одночасно
знаходяться в різних стадіях збудження і відновлення. Вони скорочуються
безладно з різною силою і амплітудою. При цьому відбувається
бомбардування атріовентрикулярного вузла величезною кількістю
імпульсів, одні з них дуже слабкі, щоб викликати збудження шлуночків,
частина застає A-V вузол в рефрактерній фазі після попереднього
скорочення. У зв'язку з цим, тільки незначна частина імпульсів досягає
шлуночків, викликаючи їх збудження і безладне скорочення. На ЕКГ
відсутні зубці P, оскільки немає систоли передсердь. Замість них
виявляються хаотичні, незначно виражені хвилі f, що мають різну форму і
реєструються з різною частотою (350-700 в одну хвилину). Ці хвилі краще
всього візуалізуются в ІІ, III стандартному відведенні і в відведеннях V1-2.
Ритм серця хаотичний, неправильний, інтервал R-R має різну тривалість,
хвилі f, нашаровуючись на зубці R, можуть злегка їх деформувати.
Комплекси QRS не змінені, мають суправентрикулярний вигляд, процеси
реполяризації не порушені. Залежно від амплітуди хвилі f розрізняють
наступні форми фібриляції передсердь: - крупнохвильову форму –
амплітуда f > 0,5 мм (при мітральному стенозі) - дрібнохвильову форму –
амплітуда f<0.5 мм (при ІБС) - нульову форму, коли хвилі f не
візуалізуються Виділяють, залежно від тривалості ФП 2 форми: 1.
пароксизмальна (персистуюча) фібриляція передсердь. 2. постійна, яка
залежно від частоти серцевого ритму розділяється на: - брадісистолічну
форму фібриляції передсердь - ЧСС<60 за одну хвилину -
нормосистолічну форму – ЧСС від 60 до 90 ударів за одну хвилину -
тахісистолічну форму – ЧСС >90 ударів за одну хвилину. 69 При
пароксизмальній формі більше скаржаться на напади серцебиття. При
постійній формі хворі можуть не відчувати перебоїв. Аускультативно –
повна хаотичність серцевих скорочень, безладність пульсу, при ЧСС, як
правило, понад 90 ударів в одну хвилину має місце дефіцит пульсу (за
рахунок безплідних скорочень). При фібриляції передсердь можуть
виникати тромбоемболічні ускладнення (особливо у хворих з
ревматичною хворобою серця). З’являються умови для формування
тромбів у вушках передсердь, внаслідок зменшення пристінкового
кровоточу по передсердям. Тріпотіння передсердь Спостерігається значно
рідше, ніж мерехтіння. При тріпотінні також відсутнє скорочення
передсердя як єдиного цілого, а є збудження і скорочення окремих
м'язових груп передсердя. Слід зазначити, що частота хвиль f при
тріпотінні значно менша, ніж при мерехтінні передсердь і складає за
звичай 220- 350 в одну хвилину, тому що ділянки м’язів які
деполяризуються значно більші ніж при мерехтінні. У A-V вузол поступає
постійне число передсердних імпульсів, у зв'язку з тим, що вузол із-за його
рефрактерності не може пропустити так багато імпульсів, виникає
функціональна атріовентрикулярна блокада. Частіше всього A-V вузол
проводить до шлуночків кожен 2 або 3 імпульс. На ЕКГ зубця P немає
(відсутня систола передсердь), спостерігаються пилкоподібні хвилі F, що
відрізняються від хвиль f при мерехтінні більшою амплітудою і меншою
частотою. Найбільш виражені хвилі f в II, III, aVF і V1 відведеннях.
Комплекс QRS не змінений (має суправентрикулярний вигляд), процеси
реполяризації не порушені. Якщо ритм шлуночків правильний і кожній
відстані R-R відповідає однакова кількість хвиль f , то говорять про
регулярну форму тріпотіння передсердь. Частіше зустрічається регулярна
форма тріпотіння передсердь 2:1 – коли проводиться кожен другий
імпульс через A-V вузол і перед кожним комплексом QRS реєструються
дві хвилі f ; рідше зустрічається форма 3:1, коли кожному QRS передує 3
хвилі f . 70 Якщо ритм неправильний, то мова йде про нерегулярну форму
тріпотіння передсердь, при якій змінюється коефіцієнт проведення 2:1, 3:1
(різна кількість хвиль f передує кожному комплексу QRS). Тріпотіння
може носити як пароксизмальний характер, так і переходити в постійну
форму. Тріпотіння і мерехтіння шлуночків При тріпотінні і мерехтінні
шлуночків відсутнє збудження шлуночків як єдиного цілого, а
відбувається скорочення окремих груп м'язових волокон. Ці стани
викликають припинення ефективної гемодинаміки, тобто зупинку
кровообігу. Дані порушення ритму є основною причиною раптової
коронарної смерті. Частота скорочень окремих м'язових волокон при
тріпотінні від 150 до 300 в одну хвилину, а при мерехтінні шлуночків від
150 до 500 в 1 хвилину. Тріпотіння шлуночків Цим терміном позначають
дуже часту ритмічну, але неефективну діяльність міокарду шлуночків. На
ЕКГ виявляється деформовані і дуже широкі пилкоподібні комплекси
QRS, сегмент S-T і зубець T, які не можна диференціювати. Шлуночкові
хвилі великі, безпосередньо перехідні одна в одну. Крива, що
реєструється, має вид синусоїди,. ізоелектрична лінія відсутня. При
тривалому часі тріпотінні переходить в мерехтіння шлуночків, яке
закінчується зупинкою серцевої діяльності, якщо не провести невідкладні
реанімаційні заходи - дефібриляцію. Мерехтіння (фібриляція) шлуночків Є
асинхронною, нерегулярною діяльністю ділянок міокарду шлуночків. Це
приводить до припинення навіть мінімального кровообігу. Виявляється на
ЕКГ повною хаотичністю, нерегулярністю і різкою деформацією
шлуночкових хвиль, які відрізняються один від одного по висоті, формі та
тривалості, а також відсутності ізолінії. Слід зазначити, що мерехтіння і
тріпотіння шлуночків мо- 71 жуть багато разів переходити один до одного.
Мерехтіння шлуночків зазвичай закінчується асистолією, і на ЕКГ
реєструється ізолінія. Тріпотіння і мерехтіння об'єднуються терміном
фібриляція шлуночків. Клінічно цей стан виявляється слабкістю,
запамороченням, через 15-20 секунд хворий знепритомніє, через 40-50
секунд з'являються судоми, мимовільне сечовипускання, пульс не
визначається, діяльність серця не вислуховується, артеріальний тиск не
визначається, спостерігається зупинка дихання хворого, а потім клінічна
смерть переходе у біологічну. Через 4 хвилини відбуваються необоротні
зміни в корі головного мозку. При появі симптомів фібриляції шлуночків
проводять реанімаційні заходи – штучне дихання, непрямий масаж серця,
дефібриляцію.
57.Артеріальна гіпертензія: визначення поняття, принципи класифікації. Первинна та вторинна артеріальна гіпертензія. Гемодинамічні варіанти артеріальної гіпертензії.
Артеріальною гіпертензією ввижають стан підвищення артеріального тиску понад 140/90 мм рт ст. (або стале підвищення артеріального тиску) Класифікують: первинну, вторинну.
Первинна характерна: підвищення артеріального тиску не може бути пов'язане з конкретним захворюванням або патологічним процесом у тих чи тих органах і системах: причина підвищення артеріального тиску залишається неясною. Для позначення цієї форми гіпертензії в різних країнах використовують два рівнозначних терміни: "есенціальна гіпертонія " і "гіпертонічна хвороба ".
Вторинна артеріальна гіпертензія: виникає як наслідок патологічних процесів у різних органах і системах.
Вона характерна для: а) захворювань нирок (гломерулонефрит, пієлонефрит, полікістоз нирок та ін.); б) пухлин надниркових залоз (феохромоцитома, альдостерома); в) уражень серця й судин (деякі вади серця, коарктація аорти); г) захворювань нервової системи (бульбарний поліомієліт, енцефаліти; травми, струси мозку та ін.). У всіх цих випадках причина гіпертензії ясна. її усунення, як правило, веде до нормалізації артеріального тиску.
Гемодинамічні варіанти артеріальної гіпертензії Оскільки величина артеріального тиску визначається загальним законом гемоди-наміки, відповідно до якого P = QR, де Р — артеріальний тиск; Q — хвилинний об'єм серця; R — загальний периферичний опір, то артеріальна гіпертензія може бути обумовлена збільшенням хвилинного об'єму серця (Q), збільшенням загального периферичного опору (R) або тим та іншим одночасно. Відповідно до цього виділяють три гемодинамічних варіанти артеріальної гіпертензії.
Гіперкінетичнш тип. Обумовлений істотним збільшенням роботи серця, у результаті чого зростає його хвилинний об'єм (Q).
Еукінетичний тип. Виникає при помірному збільшенні хвилинного об'єму серця (Q) і загального периферичного опору (R).
Гіпокінетичний тип. Його розвиток пов'язаний з істотним збільшенням загального периферичного опору (R)
58.Експериментальне моделювання АГ.
Жодна хвороба людини не має такої великої кількості різних експериментальних моделей, як артеріальна гіпертензія. За методами відтворення всі її моделі можна поділити на кілька великих груп.
I. Порушення функції центральної нервової системи. До таких відносять: а) зіткнення процесів умовного збудження та гальмування, що призводить до розвитку у тварин (собак, мавп) неврозу; б) моделювання психоемоційної напруги шляхом створення зоосоціального конфлікту (у мавп), змін біоритмів, іммобілізації тварин; в) електричну й хімічну стимуляцію лімбіч- них структур головного мозку.
II. Порушення мозкового крово- і лімфообігу. Підвищення артеріального тиску можна викликати: а) одно- і двостороннім перев’язуванням сонних і вертебральних артерій, що живлять мозок (центрально-ішемічна артеріальна гіпертензія)', б) блокадою лімфовідведення по перинев- ральних і периваскулярних лімфатичних шляхах за допомогою каоліну, що його вводять у велику цистерну мозку.
ПІ. Порушення функції депресорних регуляторних систем. Зменшення активності цих систем можна відтворювати: а) двостороннім перетинанням у кролів і собак депресорних і синусних нервів, у результаті чого знімаються гальмівні впливи з барорецепторів рефлексогенних зон дуги аорти і каротидного синуса (рефлексогенна гіпертензія, або гіпертензія розгальмування); б) центральною деаферентацією барорецепторів, що її викликають ушкодженням ядра солітарного тракту; в) пригніченням синтезу простагландинів за допомогою, наприклад, індометацину.
IV. Порушення функції нирок. Артеріальна гіпертензія ниркового походження виникає при: а) звужуванні обох ниркових артерій або звужуванні однієї ниркової артерії з видаленням другої контрлатеральної нирки (реноваскулярна гіпертензія). Виникнення гіпертензії в цьому випадку пов’язане з активацією ренін-ангіотензинової системи; б) видаленні обох нирок і переведенні тварин на гемодіаліз для запобігання уремії (рено- привна гіпертензія). її розвиток пояснюють повним припиненням депресорних функцій нирок (не утворюються ниркові простагландини та інші сполуки, що знижують тиск); в) обгортання нирок целофаном, шовком. При цьому виникає перинефрит: здавлюється ниркова паренхіма, розвивається венозний застій і гіпоксія нирок, активується ренін- ангіотензинова система.
V. Порушення гормонального стану. До артеріальної гіпертензії спричиняються: а)введення тваринам адреналіну, б) введення вазопресину, в) субтотальне видалення кори надниркових залоз. При цьому відбувається посилення регенерації залозистої тканини з підвищеною продукцією кортикостероїдів, особливо альдостерону (наднирково-регенераційна гіпертензія).
VI. Порушення водно-сольового обміну. Артеріальну гіпертензію можна моделювати: хонної солі (сольова гіпертензія); б) застосуванням мінералокортикоїдів (дезок- сикортикостерону, альдостерону) ш мінера- локортикоїдна гіпертензія', в) поєднаним введенням кухонної солі й мінералокортикоїдів.
VII. Моделі генетично обумовленої артеріальної гіпертензії. У багатьох лабораторіях світу виведено чисті лінії щурів, характерною рисою яких є гіпертензія як ознака, що спадку- ється. Це, зокрема, щури зі спонтанною гіпертензією (лінія Окамото - Аокі); щури, схильні до інсультів; новозеландські щури, міланські щури; щури, чутливі до сольової дієти, та ін.
59. Первинна артеріальна гіпертензія як мультифакторіальне захворювання. Етіологія гіпертонічної хвороби, характеристика факторів ризику.
Артеріальна гіпертензія-стале ⬆арт.
Тиску понад 160/90мм.рт.ст.
Первинна АГ-не залежить від інших хвороб, “есенціальна/гіпертензивна”.
Екзогенні фактори:
· тривала емоц напруга/хрон стрес
· ⬆ ІМТ/ожиріння+гіперінсулінемія
· Сидячий спосіб життя(гіподинамія)
· ⬆ спожив кух. солі
Ендогенні фактори(спадковість).
+Вік: ⬆АТ прямо залежить від віку і, зазвичай, АГ
з’являється у віці 30-50 років.
+Стать(чол.)
+ Тютюнопаління – нікотин та компоненти диму ⬆АТ, погіршують еластичність судин, порушують в’язкість крові, прискорюють частоту серцевих скорочень.
60.Стадії гіпертонічної хвороби та її патогенез. Роль ренін-ангіотензин-альдостеронової системи в патогенезі артеріальної гіпертензії.
Гіпертонічна хвороба – самостійне захворювання, при якому стійке підвищення АТ (вище 140 і 90 мм рт. ст. у людей молодого віку) є провідним, а іноді й єдиним симптомом хвороби.
Класифікація гіпертонічної хвороби
|
І стадія |
Відсутні об’єктивні ознаки органічних уражень. |
|
ІІ стадія |
При обстеженні виявляються хоча б одна з наступних ознак ураження органів: а)гіпертрофія лівого шлуночка; б)протеінурія (або підвищення концентрації креатиніну плазми крові до 0,177 ммоль/л); в)генералізоване або фокальне звуження артерій сітківки; г)ультразвукові або рентгенологічні дані про наявність атеросклеротичної бляшки (аорта, сонні, здухвинні або стегнові артерії); |
|
ІІІ стадія |
З’являються ознаки ураження різних органів: а)серце - лівошлуночкова недостатність, стенокардія, інфаркт міокарда; б)мозкові крововиливи (інсульти), перехідні порушення мозкового кровообігу, гіпертонічна енцефалопатія; в)очне дно - ретинальні геморагії та ексудати з набряком сосочка зорового нерва або без нього; г) нирки-рівень креатиніну плазми вище 0,177 ммоль/л, ниркова недостатність; д) судини - розшаровуюча аневризма аорти, оклюзійні ураження артерій.
|
Класифікація артеріальної гіпертензії за рівнем АТ (ВООЗ/МТГ, 1999)
|
САТ, мм.рт.ст. |
ДАТ, мм.рт.ст. |
||
|
1) |
Оптимальний |
<120 |
<80 |
|
2) |
Нормальний |
<130 |
<85 |
|
3) |
високий нормальний |
130-139 |
85-89 |
|
Гіпертензія: |
|||
|
І ступінь (м’яка АГ) |
140-159 |
90-99 |
|
|
підгрупа погранична |
140-149 |
90-94 |
|
|
ІІ ступінь (помірна АГ) |
160-179 |
100-109 |
|
|
ІІІ ступінь (тяжка АГ) |
>180 |
>110 |
|
|
Ізольована систолічна |
>140 |
<90 |
|
|
підгрупа погранична |
140-149 |
<90 |
|
Патогенез
У патогенезі гіпертонічної хвороби найбільше значення мають порушення центральних і ниркових механізмів регуляції артеріального тиску, ендотеліальна дисфункція та інші фактори. Генетичні дефекти можуть сприяти артеріальній гіпертензії внаслідок порушення різних ланцюжків регуляції артеріального тиску – трансмембранний рух іонів Na і Са, надмірне продукування пресорних речовин (або дефіцит депресорних), підвищення чутливості рецепторів до їхньої дії. Порушується механізм регуляції обміну Na, нервової регуляції артеріального тиску, ренін-ангіотензин-альдостеронової системи, змінюються реологічні властивості крові. Артеріальний тиск залежить від хвилинного об'єму серця і загального периферичного судинного опору. Артеріальний тиск підвищується при збільшенні цих параметрів або одного з них. У більшості випадків підвищення артеріального тиску спочатку зумовлене переважно збільшенням хвилинного об'єму серця. Надалі зростає загальний периферичний судинний опір. Це пов'язано з подальшим порушенням регуляції судинного тонусу. Має значення і механічне звуження просвіту артерій внаслідок підвищення вмісту в стінці судини натрію і води (що зазвичай зумовлює і підвищення її чутливості до пресорних стимулів). У тяжчих формах хвороби звуженню просвіту сприяє ураження судинної стінки (фібриноїдний некроз, васкуліт, гіпертрофія прошарків судинної стінки та атеросклероз), яке поступово підвищує загальний периферичний судинний опір. Крім того, цьому сприяє погіршання реологічних властивостей крові, підвищення її в'язкості. Усе це призводить до ішемії органів. Найчутливіші органи (органи-мішені) – це серце, мозок, нирки.
Підвищення систолічного тиску в лівому шлуночку викликає його гіпертрофію. Посилюється робота серця, що в поєднанні з його гіпертрофією підвищує потребу міокарда в кисні, а розвиток атеросклерозу коронарних артерій призводить до ІХС – стенокардії, інфаркту міокарда.
Гіпертрофія і кардіосклероз знижують скоротливу здатність міокарда і призводять до розвитку серцевої недостатності.
Ураження судин мозку веде до гіпертонічної енцефалопатії, гострого порушення мозкового кровообігу. Ураження судин нирок призводить до розвитку нефросклерозу, зниження функції нирок, хронічної ниркової недостатності
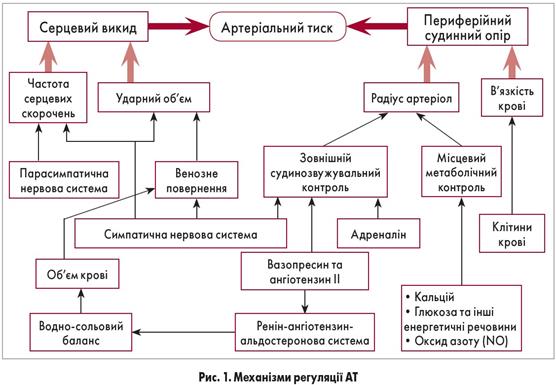
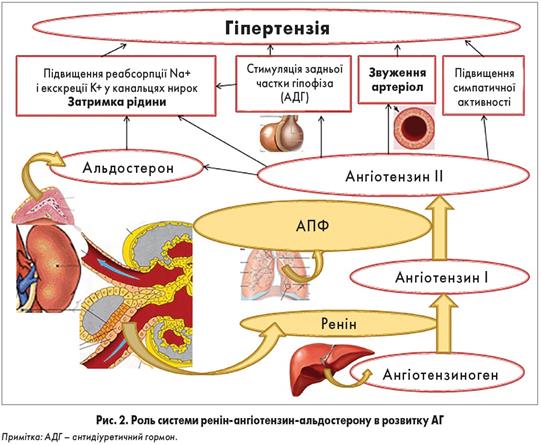
Ренін-ангіотензинна система пов'язана з функціонуванням юкстагломеруляр-ного апарату нирок (ЮГА). Цілий ряд факторів (порушення кровообігу в нирках, активація симпатоадреналової системи, зменшення концентрації іонів натрію в плазмі крові) викликає вивільнення клітинами ЮГА в кров протеолітичного ферменту — реніну (рис. 76). Ренін діє на а -глобулін плазми крові (ангіотензиноген) і відщеплює від нього пептид, що складається з десяти амінокислотних залишків. Ця речовина, яка ще не має будь-якої біологічної активності, отримала назву ангіотензин І. При проходженні через капіляри легень від ангіотензину І під впливом ферменту кон-вертази, що міститься на поверхні ендотеліальних клітин, відщеплюється дві амінокислоти, у результаті чого утворюється ангіотензин II. Далі під впливом ангіотениназ утворюються ангіотензин III (складається з 7 амінокислотних залишків) та інші пептиди, що містять 6,5 і менше амінокислот і не мають біологічної активності.
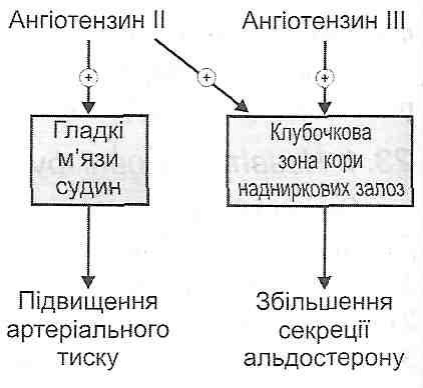
61. Причини та механізми розвитку вторинних артеріальних гіпертензій.
Ниркова гіпертензія - характеризується збільшенням показників діастолічного, або систолодіастолічного тиску. У залежності від розміру наростання діастолічного тиску розрізняють м'яку ниркову гіпертензію (90-104 мм рт. ст.), помірну (105-114 мм рт. ст.) і виражену (понад 115 мм рт. ст.). При діастолічному тиску 140 мм рт. ст. і більше, що супроводжується ураженням судин головного мозку, сітківки очей, набряком дисків очних нервів, залученням в процес нирок і серця і відсутністю вираженої позитивної динаміки при застосуванні трьох-чотирьох гіпотензивних препаратів протягом трьох місяців, гіпертензія розглядається як злоякісна НГ.
У вагітних підвищення систолічного тиску понад 30 мм рт. ст. або діастолічного понад 15 мм рт. ст. від початкового вважається прееклампсією (за умови перевищення 140/90).
Ниркова гіпертензія є наслідком цілої групи вроджених і набутих захворювань нирок та ниркових судин. Відомо, що зменшена кількість нефронів веде до розвитку гіпертензії. До найбільш вивчених і значимих варто віднести наступні патогенетичні ланки: системи, які викликають вазоконстрикцію та вазодилятацію.
Внаслідок активації ренін-ангіотензин-альдостеронової (РААС), симпатико-адреналової (САС) та вазопресин-ендотеліальної систем (ВЕС) виникає вазоконстрикція, затримка рідини в організмі, стимуляція проліферативних процесів та ремодуляція серця і судин. Основною діючою речовиною РААС і САС є ангіотензин II. Навпаки, оксид азоту, група натрійуретичних пептидів, калікріїн-кінінова система та простациклін викликають вазодилятацію, підвищення діурезу, гальмують апоптоз та ремодуляцію судин і серця. Клінічним аспектом контролю підвищеного артеріального тиску може бути блокада РААС інгібіторами АПФ і антагоністами рецепторів до ангіотензину II, САС, ВЕС та потенціювання дії вазодилятаторів.
Виділяють також гіпертензію на тлі мутації мінералкортикоїдних рецепторів. Гіпертензія характеризується супутньою гіпокаліємією та зниженим вмістом альдостерону. Відмічають підвищення тиску у вагітних на тлі підвищення прогестерону крові. Призначення спіринолактону також провокує гіпертензію у таких хворих.
Ендокринні гіпертензії обумовлені феохромоцитомою і іншими хромаффіннимі пухлинами, первинним альдостеронізмом, природженою наднирковозалозною гіперплазією з дефіцитом 11 - бета-гідроксилюванія і 17-альфа-гідроксилювания, синдромом Яценко - Кушинга, акромегалією і ін.
Лікарська АГ
У патогенезі АГ, викликаної ЛЗ, можуть мати значення вазоконстрикция через стимуляції симпатоадреналової системи або прямого впливу на клітини гладеньких м'язів кровоносних судин, збільшення в'язкості крові, стимуляція ренін-альдостерон-ангіотензинової системи (РААС), затримка іонів натрію і води, взаємодія з центральними регуляторними механізмами.
алкогольна АГ
В 5-25% випадків причиною АГ є хронічне вживання алкоголю. Точний механізм гіпертензивної дії алкоголю не відомий. Обговорюється значення стимуляції симпатичної НС, збільшення продукції глюкокортикоїдів, гіперінсулінемії, збільшення захоплення іонів кальцію клітинами і підвищення ОПСС під впливом алкоголю.
Причини вторинної АГ:
1) хвороби нирок:
a) паренхіматозні (ренопаренхіматозна АГ)
б) судинні (вазоренальна [реноваскулярна] АГ)
в) ренін-продукуючі пухлини, які походять з юкстагломерулярного комплексу нирок;
г) синдроми первинної ретенції натрію — синдром Лідла, синдром Гордона;
2) хвороби залоз внутрішньої секреції — первинний гіперальдостеронізм, синдром Іценка-Кушинга, феохромоцитома і парагангліоми, гіпер- або гіпотиреоз, гіперпаратиреоз, карциноїдний синдром, акромегалія;
3) коарктація аорти;
4) прееклампсія або еклампсія;
5) гострий стрес — опіки, алкогольний абстинентний синдром, психогенна гіпервентиляція, гіпоглікемія, стан після великих хірургічних втручань;
6) синдром обструктивного апное сну (СОАС);
7) збільшений об'єм внутрішньосудинної рідини;
8) хвороби нервової системи — підвищення внутрішньочерепного тиску, синдром Гійєна-Барре, тетраплегія, сімейна дизавтономія;
9) ЛЗ — симпатоміметики (також у формі назальних крапель), глюкокортикоїди, еритропоетин, циклоспорин, такролімус, інгібітори МАО, НПЗП, препарати солодки, карбеноксолон, пероральні контрацептиви;
10) токсичні речовини — наркотики (амфетамін, кокаїн), важкі метали, алкоголь, нікотин.
№62. Артеріальна гіпотензія. Етіологія та патогенез гострих і хронічних артеріальних гіпотензій. Колапс.
· Артеріальна гіпотензія – це стан, що характеризується зниженням систолічного тиску нижче 100 мм рт.ст., діастолічного — нижче 60 мм рт.ст. Іноді цей стан може бути в основі самостійної хвороби, також може бути симптомом багатьох захворювань.
Артеріальна гіпертензія поділяються на первинну та вторинну в свою чергу вторинна буває гостра і хронічна.
Етіологія
Вторинну АГТ можуть спричиняти запальні та дистрофічні захворювання серця і судин, тубулопатії, хвороби крові. Внаслідок патологічних процесів можливе зниження серцевого викиду, загального периферичного опору судин, зменшення ОЦК, в’язкості крові, що спричиняє розвиток гіпотензії. Здебільшого у пацієнтів із вторинною АГТ симптоматика основного захворювання превалює, проте у деяких випадках АГТ є раннім проявом цього захворювання . Хронічна АГТ виникає на тлі анемії, голодування, гіповітамінозу, вираженого астенічного синдрому.
Патогенез
АТ регулює низка механізмів, які тісно взаємопов’язані. Відповідно до гідравлічного рівняння на рівень системного АТ прямий вплив має серцевий викид і периферичний опір судин. Ці два фактори забезпечують контроль АТ на трьох основних анатомічних ділянках: артеріоли, венули та серце. Нирки, підтримуючи об’єм внутрішньосудинної рідини, також беруть участь у контролі АТ.
· Колапс – це гостра судинна недостатність,котра характеризується різким падінням артеріального і венозного тиску й зменшенням маси циркулюючої в судинній системі крові.
№63. Артеріосклероз: визначення поняття, класифікація. Загальна характеристика різних форм артеріосклерозу (типова локалізація, прояви, ускладнення).
· Артеріосклероз - захворювання, яке характеризується потовщенням стінок артерій, скороченням їх просвіту, що призводить до обмеження припливу крові до органів і тканин. Здоровим артеріях властива гнучкість і еластичність, з розвитком захворювання на стінках утворюються бляшки, відбувається склеротірованіе артерій.
Атеросклероз це одна з форм артеріосклероз.
Залежно від переваги локалізації атеросклерозу в тому чи іншому судинному басейні виділяють:
· Атеросклероз аорти — найчастіша клінічна форма. Процес може тривало перебігати безсимптомно. Значно виражений в її черевному відділі, характеризується атероматозом, виразкуванням і петрифікацією. Часто ускладнюється тромбозом, емболією з розвитком інфарктів нирок і гангреною кишківника, нижніх кінцівок, розшаровуючою аневризмою аорти.
· Атеросклероз коронарних артерій лежить в основі розвитку ішемічної хвороби серця.
· Атеросклероз артерій головного мозку є основою цереброваскулярного захворювання. Тривала ішемія головного мозку внаслідок стенозуючого атеросклерозу призводить до енцефалопатії .
· &nnbsp; При атеросклерозі ниркових артерій виникають клиноподібні ділянки атрофії паренхіми з колапсом строми, або інфаркти з наступною їх організацією і формуванням втягнутих рубців .
· Атеросклероз артерій кишечника, ускладнений тромбозом, здебільшого закінчується гангреною кишок.
· При атеросклерозі артерій нижніх кінцівок частіше уражаються артерії стегна, розвивається атрофія м'язів, поява характерного болю під час ходи . Ускладнюється гангреною нижньої кінцівки
№64. Атеросклероз. Етіологія атеросклерозу: фактори ризику, причинні фактори. Роль пошкодження ендотелію на начальних етапах патологічного процесу.
· Атеріосклероз – хронічне захворювання, що уражає переважно великі артеріальні судини; здебільшого спостерігається у людей похилого віку.
Виникненню атеросклерозу сприяє багато факторів: підвищення кров'яного (артеріального) тиску, ангіоспазм, який спричинюють розлади нервової регуляції судин, нервово-психічні фактори, порушення діяльності ендокринних залоз.
Розвиток атеросклерозу пов'язаний із загальними порушеннями обміну ліпідів; при цьому в крові протягом тривалого часу (або періодично) збільшується вміст холестерину, особливо фракцій ліпопротеїнів низької щільності та дуже низької щільності. Це спричинює відкладання холестерину та інших ліпідів у внутрішню оболонку стінок артерій з подальшою глибокою зміною їхньої будови.
№65. Експериментальне моделювання атеросклерозу. Патогенетичне значення дисліпідемії.
Дисліпідемія – включає підвищення рівня ліпідів і ліпопротеїнів вище оптимального значення та/або можливе зниження показників частини ліпідного спектра, а саме ЛПВЩ або альфа-ліпопротеїнів.
№66. Стадії та патогенез атеросклерозу. Теорії атерогенезу. Стадійність клітинних реакцій в процесі розвитку метазапалення при атеросклерозі.
Патогенез атеросклерозу називають атерогенез. Він відбувається в кілька етапів.
Розвиток атеросклеротичного ураження - це сукупність процесів надходження в інтиму і виходу з неї ліпопротеїдів і лейкоцитів, проліферації і загибелі клітин, утворення й перебудови міжклітинної речовини, а також розростання судин і звапнення. Ці процеси управляються безліччю сигналів, часто різноспрямованих. Накопичується все більше даних про складну патогенетичного зв'язку між зміною функції клітин судинної стінки і мігрували в неї лейкоцитів і чинниками ризику атеросклерозу.
67. Роль пошкодження ендотеліальної дисфункції в атерогенезі. Фактори, що викликають ушкодження ендотелію. Роль регуляторних ендотеліальних факторів (тромбоксану, молекул міжклітинної адгезії, цитокінів).
І. Стадія доліпідних змін в основі лежить ендотеліальна дисфункція
Ушк. ендотелію суд. може бути обумовлене:
а) мех.. і фіз. факторами (тиск крові, її турбулентний рух, іонізуюча радіація);
б) хім.. спол. (нікотин, гомоцисте- їн, великі дози вітаміну D);
в) ендотеліотропними вірусами, токс. Бакт. (веротоксин);
г) імен. факторами (комплекси “антиген - антитіло”).
Причини ендотеліальної дисфункції :
а) вільні радикали і пероксиди, що утв. у вел кільк в умовах оксидаційного стресу
б) окиснені ЛПНГ
в) продукти неферментативного глікозилю- вання білків (AGEs), що утвор. при цукровому діабеті внаслідок тривалої гіперглікемії
г) характерні для гемодинаміки фізичні фактори, зокрема напруга зсуву;
д) інфекційні агенти: бактеріальні токсини, віруси;
Ендотеліальна дисфункція виявляє себе змінами властивостей і діяльності ендотеліоцитів:
1) ⬆проникність ендотелію ліпопротеїнів;
2) ⬆адгезивність ендотелію до лейкоцитів крові та тромбоцитів. Це пов’язано з появою на люмінальній поверхні ен- дотеліоцитів молекул клітинної адгезії-. Р- та Е-селектинів, подібних до імуноглобу- лінів білків (ІСАМ-1, ІСАМ-2, VCAM-1), здатних взаємодіяти з адгезивними молекулами (олігосахаридами, інтегринами) лейкоцитів
3) посилюється утвор.прокоагулянтних і пригнічується синтез антикоагулянтних факторів, що порушує баланс між цими системами гемостазу і створює сприятливі умови для тромбоутворення
4) ⬆ синтез великої кількості біологічно активних сполук (цитокіни, фактори росту, вазоактивні речовини )
68. Значення порушень вмісту в крові основних фракцій ліпопротеїнів. Роль спадкових та набутих порушень рецептор-опосередкованого транспорту ліпопротеїнів (ЛП) (порушення рецепторів ЛП, модифікація ЛП) в атерогенезі.
а) ⬆ вміст в плазмі крові “атерогенних” ліпопротеїдів низької (ЛПНГ) і дуже низької густини (ЛПДНГ);
б) появою в крові не властивих для норми ліпопротеїдів "модифікованих". (глікозильовані, ацетоацетильовані ліпопротеїди, ліпопротеїди, зв'язані з продуктами ПОЛ; комплекси ліпопротеїд-антитіло та ін.;)
в) ⬇вмісту в плазмі крові ліпопротеїдів високої густини (ЛПВГ- “антиатерогенні”).
69. Патогенез синдрому відкритої артеріальної протоки у новонароджених. Принципи лікування
Відкрита артеріальна (Боталова) протока – незарощена судина, яка з’єднує аорту з легеневою артерією.
Патогенез:Незаростання боталової протоки ➡кров з аорти викидається в легеневу артерію( оскільки тиск в аорті істотно вищий) ➡ в малому колі кровообігу сильно ⬆об’єм крові = 1) основна частина крові залишається в легенях, а інші органи і системи постійно недоотримують кров, перебуваючи в стані кисневого голодування, 2)Внаслідок підвищеного тиску в судинах легенів виникають застійні явища, що сприяє розвитку запального процесу ➡ судини склерозируются, а їх функціональність порушується.
Лікування: діагноз є абсолютним показанням для проведення хірургічної операції.Ідеальним віком для проведення операції вважається 3-5 років.
70.
Особливості
та механізми змін гемодинаміки новонародженого при переході від фетального
кровообігу до неонатального.
ОСОБЛИВОСТІ
КРОВООБІГУ ПЛОДА
• Плацентарний кровообіг представлений 2 пупковими артеріями та пупковою веною, у складі пупкового канатика
• Починає забезпечувати газообмін плода з 2 міс
• Мале коло кровообігу не функціонує
• Всі органи плода отримують змішану кров, але до життєво важливих органів (мозок, серце, печінка) надходить макс оксигенована кров з висхідної аорти та її дуги
• Наявність фетальних шляхів кровообігу та шунтів (аранцієва і Боталова протока, овальний отвір та ін)
• Низький кров’яний тиск у легеневій артерії й аорті
Таким чином, система кровообігу плода становить собою замкнуте коло, відокремлене від системи кровообігу матері, і діє винятково за рахунок скоротливої здатності серця плода. З 11-12- го тижня певну допомогу надають дих. рухи (періоди від’ємного тиску). ЧСС ембріону спочатку низька (15-35/хв), потім збільшується до 125-130/хв.
ОСОБЛИВОСТІ КРОВООБІГУ НОВОНАРОДЖЕНОГО
• Припиняється плацентарний кровообіг.
• Різко збільшується кровоплин через легеневі судини, знижується опір в легеневій артерії через вазодилятацію легеневого русла.
• Переключаються насоси правого і лівого серця з паралельно працюючих на послідовно включені.
• Відмічаються фізіол тахікардія (ЧСС 140-160 уд/хв) та низький АТ(60-70 мм рт.ст.).
• Підвищується потреба у кисні, зростають серцевий викид і системний судинний тиск.
• Закриваються фетальні судинні комунікації: - венозна (аранцієва) протока спустошується з перших хвилин життя, повна облітерація закінчується наприкінці 8-го тижня життя; - спазм артеріальної (Боталової) протоки настає з першим вдихом, але вона функціонує ще протягом 8 годин (тому у 85% новонароджених вислуховуються серцеві шуми). Анатомічна облітерація – до 2 місяців; - овальний отвір заростає наприкінці 5-7 місяця життя.
71. Фактори ризику розвитку атеросклерозу у дітей та підлітків. Спадкові форми атеросклерозу. Сімейна гіперхолестеринемія: які спадкові дефекти, гомо- та гетерозиготні форми СГХС, клінічні прояви, діагностика, принципи лікування.
Фактори ризику атеросклерозу у дітей:
· шкідливі звички, ожиріння у дітей
· малорухливий спосіб життя
· неправильне харчування і стреси.
· Якщо родичі хворіли на ішемічну хворобу або інше захворювання такого ж типу.
· надлишкова вага, гіпертонія і цукровий діабет у дитини. Якщо у батьків рівень холестерину перевищує 5,2 ммоль на літр, ймовірність розвитку цього захворювання в дитини суттєво зростає.
Сімейна гіперхолестеринемія (СГ) – найбільш поширене домінантно успадковане захворювання людини , яке найчастіше викликається домінантною мутацією втрати функції в гені, що впливає на ліпопротеїни низької щільності (ЛПНЩ), які видаляють ЛПНЩ з плазми крові ,у результаті чого з народження рівень холестерину ЛПНЩ значно підвищується.
Клін. критерії гетерозиготної сімейної гіперхолестеринемії: вис. рівень аг. холестерину і ЛПНЩ плазми крові, сімейний анамнез гіперхолистеринемії (ГХС) (особливо випадки ГХС у дітей), відкладення холестерину в екстраваскулярні ткан. у вигляді ксантом сухожиль або ліпоїдна дуга рогівки і ранній розвиток ІХС у пацієнта чи його родичів.
Спец. діагностичною ознакою СГ є ксантоми сухожиль. Найбільш частою локалізацією ксантом є ахіллове сухожилля і сухожилля-розгиначів пальців кистей (в області п’ястно-фалангових суглобів). При ретельному розпитуванні у хворих можна виявити в анамнезі тендовагініти.
Дуга рогівки і ксантелазм специфічні для гетерозиготної СГ у осіб молодше 45 років, а в похилому віці - поруш. ліп. обміну.
СГ ➡ ІХС(атеросклероз коронарних артерій)у більш ранньому віці, ніж можна було б очікувати в загальній популяції ➡стенокардії (стиснення в грудях при фізичному навантаженні)/ серцевого нападу.
СГ ➡ураж. Суд. ГМ➡ транзиторна ішемічна атака (короткі епізоди слабкості в одній стороні тіла або нездатність говорити)/ ішемічного інсульту.
Терапія СГ складається з двох компонентів:
· немедикаментозного (дієти, регулярну фіз..акт., повне припинення куріння і підтримання маси тіла на оптимальному рівні
· медикаментозного (для зниження ХС ЛПНЩ і досягнення його оптимального рівня):статини (інгібітори гідроксиметилглутаріл-коензим А редуктази – ферменту, що регулює швидкість біосинтезу холестерину з ацетату), секвестранти жовчних кислот (іонообмінні смоли) та езетіміб – селективний інгібітор абсорбції холестерину в стінці тонкого кишечника. Інші гіполіпідемічні засоби (нікотинова кислота і фібрати) слід призначати додатково до терапії статинами тільки у випадках супутньої гіпертригліцеридемії.)
72.Фагоцитоз.Стадії та їх механізми.Роль фагоцитозу при запаленні.Порушення фагоцитозу та їх прояви у дитячому віці.
Фагоцитоз— активне захоплення і поглинання мікроскопічних сторонніх об'єктів (бактерії, фрагменти клітин) і твердих частинок одноклітинними організмами або деякими клітинами багатоклітинних тварин.
Механізм фагоцитозу. Розрізняють чотири стадії фагоцитозу: 1. Зближення. 2. Прилипання. 3. Поглинання. 4. Перетравлення.
У процесі фагоцитозу активна роль належить клітинній мембрані, яка обволікає часточку, що фагоцитується, і втягує її вглибину цитоплазми з утворенням фагосоми. З лізосом клітини до фагосом надходять гідролітичні ферменти, які перетравлюють поглинуту часточку. Неперетравлені рештки можуть лишатись у клітині довгий час. Фагоцитоз відіграє важливу роль, головним чином, при запаленні, загоєнні ран, як фактор неспецифічного імунітету.
Порушення фагоцитозу.Активність фагоцитозу може знижуватись внаслідок розладу утворення фагоцитів і пригнічення їх активності.Ці порушення можуть бути спадковими та набутими.
Зниження активності фагоцитозу відбувається пів впливом гормонів-глюкокортикоїдів,медіатора нервової сстеми-ацетилхоліну,у разі нестачі в організмі певних гормонів,вітамінів,порушення водно-мінерального балансу.Активність фагоцитозу може знижуватися під впливом гліколітичної отрути-монойодацетату,який порушує вироблення енергії в лейкоцитах,бактеріальних токсинів,що призводить до незавершеного фагоцитозу.Фагоцитарна активність деяких ферментів у лейкоцитах різко знижена при лейкозі,імунній недостатності та дефіциті комплементу,оскільки в нормі антитіла й комплемент стимулюють фагоцитоз.
73.Проліферація клітин в осередку запалення,її стадії та механізми.Механізми міогенної дії факторів росту і цитокінів.Регенерація та фіброплазія як способи загоєння.Особливості проліферативного запалення у дитячому віці(на прикладі утворення туберкульоми)
Проліферація є завершальною фазою розвитку запалення, що забезпечує репаративні регенерацію тканин на місці вогнища альтерації.
Проліферація розвивається з самого початку запалення поряд з явищами альтерації і ексудації.
Розмноження клітинних елементів починається по периферії зони запалення, в той час як в центрі вогнища можуть ще прогресувати явища альтерації і некрозу.
Повного розвитку проліферація сполучнотканинних і органоспецифическихклеточных елементів досягає після "очищення" зони ушкодження від клітинного детриту та інфекційних збудників запалення тканинними макрофагами і нейтрофілами. У зв'язку з цим слід зазначити, що процесу проліферації передує утворення нейтрофільного і моноцитарного бар'єрів, які формуються по периферії зони альтерації.
Відновлення і заміщення пошкоджених тканин починається з виходу з судин молекул фібриногену та освіти фібрину, який формує своєрідну сітку, каркас для подальшого клітинного розмноження. Вже з цього каркасу розподіляються в осередку репарації швидко утворюються фібробласти.
Розподіл, зростання і переміщення фібробластів можливо тільки після їх зв'язування з фібрином або колагеновими волокнами. Цей зв'язок забезпечується особливим білком - фібронектином.
Регенераці-це проліферація попередників паренхіматозних клітин,що залишилися.У цьому випадку відбувається заміна втрачених клітин клітинами того самого типу.Регенерація характернка для для тканин,клітин яких постійно проліферують і тканин,що в нормі мають низький рівень реплікації,але за необхдності під впливом відповідних факторів росту можуть розпочати компенсаторну гіперплазію.
74.Роль реактивності організму в розвитку запалення.Особливості запалення у дтячому віці.Зв*язок між патологічною імунною відповіддю і запаленням.Вплив гормональних чинників на запалення.
Реактивність — це властивість організму і його структур відповідати змінами життєдіяльності на дію факторів зовнішнього середовища.Реактивність забезпечує взаємодію організму з навколишнім світом. Вона істотно впливає на розвиток і перебіг хвороб.
Фактори:1. Вік. Для раннього дитячого віку характерна низька реактивність. Вона поступово зростає, стає максимальною у дорослих, а в літньому віці починає зменшуватися.
2. Стать,
3. Спадковість.
4. Конституція (див. розд. 6).
5. Функціональний стан нервової, ендокринної, імунної систем і сполучної тканини.
6. Фактори навколишнього середовища (клімат, характер харчування, соціальні умови та ін.).
По механізму впливу на запалення гормони ділять на прозапальних і протизапальних. Прикладом провоспалітел'них гормонів, тобто гормонів, що підсилюють запалення, можуть бути мінералокортикоїди, зокрема, Альдостерон. Під його впливом істотно підвищується проникність стінки судин, внаслідок чого збільшується ексудація. Найбільш важливими протизапальними гормонами є глюкокортикоїди. У високих (лікувальних) дозах вони викликають дерепресію гена, що кодує структуру особливого білка, — ліпокортіна. Ліпокортін є природним внутріклітинним інгібітором фосфоліпази А2. Пригноблення цього ферменту ліпокортіном має два важливі слідства: 1) з одного боку, зменшується утворення лізофосфоліпідов, внаслідок чого зменшуються явища альтерації (стабілізація мембрани); 2) з іншого боку, зменшується утворення арахидонової кислоти і її похідних (простагландінов, тромбоксанов, простациклінов, лейкотрієнів). Ця обставина пояснює інші протизапальні ефекти глюкокортикоїдів — зменшення ексудації і еміграції лейкоцитів, ослабіння порушень мікроциркуляції. В результаті пригноблення процесів клітинного ділення і біосинтезу білка зменшуються явища проліферації у вогнищі запалення.
75.Гарячка:визначення поняття,принципи класифікації.Зв*язок між гарячко. І запаленням.Види пірогенів.Утворення пірогенів при інфекціїї,асептичному ушкодженні та імунних реакціях.Хімічна природа і походження вторинних пірогенів,механізм дії.
Гарячка- типовий патологічний процес, який виявл. підвищ. температури тіла у відповідь на дію пірогенних факторів.Розрізняютьінфекційні (як відповідь на проникнення в організм мікроорганізмів та їх токсинів) й неінфекційні (при потрапл. в організм реч., які не мають віднош. до інфекції) причини гарячки.Пірогенниминазив. реч, які потрапл. в організм ззовні або утв. всередині, спричинюють гарячку.За походж.:екзогенні й ендогенні, за механізмом дії: первинні і вторинні. Під впливом первинних пірогенів утв. вторинні. Цей процес відбув. у макрофагах та у нейтрофільних гранулоцитах. Механізм: разом із збудником в організм потрапляють пірогени-бактеріальні токсини. вони вплив. на макрофаги й нейтрофілільні гранулоцити, які почин. синтезувати Іл 1. Циркулюючий у крові Іл 1 діє на клітини-мішені. Під впливом Іл 1 на рівні мозкових артеріол і капілярів утв. простагландини Е1 і Е2,які проходять крізь бар’єр і впливають на центр терморегуляції.
76.Гарячка:стадії розвитку,зміни терморегуляції,обміну речовин та фізіологічних функцій.Захисне значення та патологічні прояви гарячки.Принципи жарознижувальної теріпіїї.Поняття про піротерапію.
1.Стадія підвищення температури: тепловіддача зменш.внаслідок звуж. периферичних судин і зменш. припливу крові до тканин. Теплопродукція збільш. за рахунок скорочувального термогенезу,розєднання окисного фосфорування і окиснення.2.Стадія стояння температури на підвищеному рівні:тепловіддача збільш. за рахунок розшир. периферичних судин.,теплопрод. не змін.3.Стадія зниження температури:зниж. теплопродукції,тепловід. не змін. Температурна крива склад. з 3 частин:підвищення,стояння,зниження.;залеж. Від збудника,організму хворого,від здатн. Імунної системи відповідати на антигенні стимули.Основні типи температурних кривих:1)febrisintermittens-температура нормалізується один або декілька раз на добу(гнійна інфекція,абсцеси,туберкульоз);2)febrisremittens-зміна температури являє більше 1 С на добу,але вона не повертається до норми(вірусні,бактеріальні інфекції);3)febris continua-добові зміни температури являють менше 1 С(черевний и сипний тиф);4)febris recurrens-напади підвищення температури чергуються з періодами її нормалізації,що продовжуються декілька діб(поворотний тиф,малярія).
Нормальний перебіг обміну енергії на молекулярному рівні зумовлює динамічну взаємодію процесів катаболізму і анаболізму.У разі порушення катаболічних процесів передусім страждає регенерація АТФ,а також надходження субстратів,потрібних для біосинтезу(анаболізму). Порушення анаболічних процесів призводить до розладу відтворення функціонально важливих сполук (ферментів,гормонів), потрібних для катаболізму. Щоб визначити поруш. обміну реч., виходять з основного обміну, на який впливають різні фактори: рефлекторні, умовнорефлекторні, гормональні. Особливо чітко це виявл. в умовах патології, коли поруш. нейрогормональна регуляція обміну . Підвищення ОО на 20% є ознакою тиреотоксикозу, а зниження про гіпофункцію щит. залози. Гіпофункція гіпофіза, призводить до зниж. теплопродукції й основного обміну. У разі гіпофункції внутр. стат. органів інтенсивність енерг. процесів і ОО зниж.
Механізми дії антипірогенної терапії є достатньо різноманітними, що дозволяє клініцисту вибрати у кожному конкретному випадку найбільш адекватні засоби.
При застосуванні етіотропної терапії лікар позбавляє організм від дії первинних пірогенів і таким чином, переривається ланцюжок, що призводить до утворення вторинних пірогенів, а отже, гарячка зникає. Але часто цей принцип є недосяжним, тому найдієвішим є патогенетичний принцип лікування гарячок. В його основі лежить зниження «установочної точки» центру терморегуляції. Це досягається пригніченням утворення простагландинів групи Е за допомогою застосування інгібіторів циклооксигенази. До них належать ацетилсаліцилова кислота, індометацин, парацетамол та інші ненаркотичні анальгетики. Ще один шлях пригнічення утворення простагландинів Е – застосування інгібіторів фосфоліпази А2 .
77. Порушення регуляції зовнішнього дихання. Брадипное, тахіпное, гіперпное, апное.
Недостатність зовнішнього дихання — це патологічний стан, при якому система зовнішнього дихання не здатна забезпечити нормальний склад газів крові (газовий гомеостаз).
При патології внаслідок дії рефлекторних, гуморальних або інших чинників на дихальний центр можуть змінюватися ритм, глибина і частота дихання, а також може виникати задишка. Ці зміни можуть бути проявом компенсаторних реакцій організму, спрямованих на підтримку сталості газового складу крові, або проявом порушень нормальної регуляції дихання, що ведуть до розладів альвеолярної вентиляції, до недостатності дихання.
Зміни центральної регуляції дихання можуть виявляти себе такими його типами. 1. Брадипное — рідке дихання. Механізм розвитку рідкого дихання полягає в зміні характеру нервової імпульсації, що йде від різних рецепторів до дихального центру, або в первинному порушенні діяльності самих дихальних нейронів. Рефлекторне зменшення частоти дихання може спостерігатися при підвищенні артеріального тиску (рефлекс із барорецепторів дуги аорти і каротидного синуса), при гіпероксії (унаслідок періодичного збудження хеморецепторів, чутливих до зниження напруги кисню в артеріальній крові).
Глибоке рідке дихання може з'явитися при підвищенні опору руху повітря у верхніх дихальних шляхах — стенотичне дихання. У цьому випадку вдих і видих відбуваються повільніше, ніж звичайно. У встановленні такого типу дихання певну роль відіграють імпульси, що надходять у дихальний центр від міжреберних м'я-. зів, що працюють з підвищеним навантаженням. Крім того, має значення запізнювання в цьому випадку гальмівного рефлексу Герінга-Брейєра. Брадипное може розвиватися в результаті безпосередньої дії патогенних факторів на дихальний центр, що знижує збудливість дихальних нейронів. Пригнічення дихального центру можливе при тривалій і тяжкій гіпоксії (в умовах розрідженої атмосфери, при недостатності кровообігу та ін.), за умов впливу речовин з наркотичною дією, при деяких органічних ураженнях головного мозку (запалення, порушення мозкового кровообігу, набряк та ін.) і функціональних розладах центральної нервової системи (невроз, істеричні реакції). У всіх цих випадках рідке дихання може супроводжуватися зменшенням його глибини, що призводить до зниження альвеолярної вентиляції і розвитку недостатності дихання. 2. Поліпное (тахіпное) — часте поверхневе дихання. В основі розвитку поліпное лежить рефлекторна перебудова роботи дихального центру. У деяких тварин (наприклад, у собак) часте поверхневе дихання виникає при дії високої температури. У людини поліпное може спостерігатися при гарячці, функціональних порушеннях центральної нервової системи (істерія), ураженні легень (ателектаз, пневмонія, застійні явища).
Крім того, до розвитку поліпное може спричинятися біль, що локалізується у ділянках тіла, які беруть участь у дихальному акті (грудна клітка, черевна стінка, плевра). Біль призводить до обмеження глибини дихання і збільшення його частоти (щадне дихання). Поліпное знижує ефективність дихання, тому що при цьому значно зменшується альвеолярна вентиляція і газообмін в основному відбувається у "мертвому" просторі. 3. Гіперппое — глибоке часте дихання. В умовах патології гіперпное розвивається при інтенсивній рефлекторній або гуморальній стимуляції дихального центру, наприклад, при зниженні парціального тиску кисню у вдихуваному повітрі або при підвищенні в ньому концентрації С02, при анемії, ацидозі і т. д. Крайній ступінь збудження дихального центру виявляється диханням Куссмауля, яке найчастіше спостерігається у хворих у стані діабетичної коми. Воно являє собою гучне часте дихання, при якому після глибокого вдиху настає посилений видих з активною участю експіраторних м'язів. 4. Апное— тимчасова зупинка дихання. Тимчасова зупинка дихання може бути пов'язана зі зменшенням рефлекторної або безпосередньої хімічної стимуляції дихального центру. Наприклад, апное виникає у тварини або людини після пасивної гіпервентиляції під наркозом унаслідок зменшення в артеріальній крові напруги CO і припиняється зразу ж, як тільки вміст С02 нормалізується. При швидкому підвищенні артеріального тиску, наприклад при введенні в кров адреналіну, також, спостерігається апное (рефлекс із барорецепторів). Апное, що часто повторюється і порушує звичайний ритм дихання, може бути пов'язане зі зниженням збудливості нейронів дихального центру (гіпоксія, інтоксикація, органічні ураження головного мозку).
78. Періодичне дихання, термінальне дихання. Етіологія, патогенез.
Періодичним диханням називають таке порушення ритму дихання, при якому періоди дихання чергуються з періодами апное. Найчастіше бувають два типи періодичного дихання: дихання Чейна-Стокса і дихання Біота. Дихання Чейна—Стокса характеризується наростанням амплітуди дихання до вираженого гіперпное, а потім зменшенням її до апное, після якого знову настає цикл дихальних рухів, що закінчуються також апное.
Циклічні зміни дихання у людини можуть супроводжуватися потьмаренням свідомості в період апное і його нормалізацією в період збільшення вентиляції. Артеріальний тиск при цьому також коливається, як правило, підвищуючись у фазі посилення дихання і знижуючись у фазі його ослаблення. У більшості випадків дихання Чейна-Стокса є ознакою гіпоксії головного мозку. Воно може виникати при недостатності серця, захворюваннях мозку і його оболонок, уремії. Деякі лікарські препарати (наприклад, морфін) також можуть викликати дихання Чейна — Стокса. Його можна спостерігати і у здорових людей на великій висоті (особливо під час сну), у недоношених дітей, що, очевидно, пов'язане з недосконалістю нервових центрів. Патогенез дихання Чейна-Стокса можна представити в такий спосіб. Клітини кори великого мозку і підкіркових утворень унаслідок гіпоксії пригнічуються — дихання зупиняється, свідомість зникає, пригнічується діяльність судинорухового центру. Однак хеморецептори при цьому все ще здатні реагувати на зміни вмісту газів у крові. Різкого посилення імігульсації з хеморецепторів, поряд із прямою дією на центри високої концентрації вуглекислого газу і стимулами з барорецепторів унаслідок зниження артеріального тиску, виявляється достатньо для збудження дихального центру — дихання поновлюється. Поновлення дихання веде до оксигенації крові, що зменшує гіпоксію головного мозку і поліпшує функцію нейронів судинорухового центру. Дихання стає глибше, свідомість проясняється, підвищується артеріальний тиск, поліпшується наповнення серця. Вентиляція, що збільшується, веде до підвищення напруги кисню і зниження напруги С02 в артеріальній крові. Це у свою чергу призводить до ослаблення рефлекторної і хімічної стимуляції дихального центру, діяльність якого починає вгасати, - настає апное. Дихання Біота відрізняється від дихання Чейна-Стокса тим, що дихальні рухи, які характеризуються постійною амплітудою, раптово припиняються, так само як і раптово починаються. Найчастіше дихання Біота спостерігається при менінгіті, енцефаліті та інших захворюваннях, що супроводжуються ушкодженням центральної нервової системи, особливо довгастого мозку.
Термінальним називають дихання, що виникає в термінальних станах, тобто за умов, коли організм перебуває на межі життя і смерті. Найчастіше реєструють два типи термінального дихання: апнейстичне і гаспінг-дихання.
Апнейстичие дихання характеризується конвульсивним зусиллям вдихнути, що не припиняється, але зрідка переривається видихом. В експерименті його спостерігають після перетинання у тварини обох блукаючих нервів і мозкового стовбура між пневмотаксичним (у ростральній частині мосту) і апнейстичним (у середній і каудальній частинах мосту) центрами (рис. 136). Вважають, що апнейстичний центр має здатність збуджувати інспіраторні нейрони, які періодично гальмуються імпульсами із блукаючого нерва і пневмотаксичного центру. Перетинання зазначених структур призводить до постійної інспіраторної активності апнейстичного центру.
Гаспінг-дихання - це поодинокі, рідкі вдихи, частота і амплітуда яких поступово зменшується аж до повної зупинки дихання. Такий в ид дихання спостерігають при агонії, наприклад у заключній стадії асфіксії. Звичайно характерні для гаспінг- дихання рідкі і низькоамплітуцні вдихи виникають після тимчасової зупинки дихання (претермінальної паузи). Поява їх, можливо, пов'язана зі збудженням клітин, що містяться в каудальній частині довгастого мозку, після вимикання функції вище розташованих відділів мозку.
Кардіогенні механізми танатогенеза при СРДС пере дусім асоціюються з розвитком життєнебезпечних арит мій. Провідна роль надається двом основним гіпотезам: гіпотезі симпатичного дисбалансу і, як наслідок, подов ження інтервалу QT з можливим розвитком електричної нестабільністі міокарда та гіпотезі про генетично детермі новані летальні серцеві каналопатії, наявність яких перед бачає виникнення життєво небезпечних порушень серце вого ритму через порушення роботи трансмембранних іонселективних каналів клітин міокарда.
Респіраторний механізм розвитку СРДС включає декілька етапів: 1. Життєнебезпечна подія, що виникає у сні (напри клад, спазм дихальних шляхів, гіперкапнія, апное внаслі док ларингеального хеморефлексу або гастроезофагеаль ної регургітації), призводить до гострої асфіксії і гіпопер фузії мозку. Асфіксія є основним стресорним фактором. Ситуація посилюється при високій температурі навко лишнього середовища з розвитком гіпертермії і порушен ням серцеволегеневої регуляції [51]. 2. У нормі у відповідь на гостру гіпоксію і гіперкапнію у сні виникає захисна arousalреакція, яка передбачає фор соване дихання (виникнення «гіпоксичної» задишки), актив не розширення верхніх дихальних шляхів і пробудження. Аrousalреакція формується в результаті послідовної специфічної активації корковопідкоркових структур головного мозку і включає активуючі і гальмуючі компоненти, які здій снюють взаємодію між корковими і підкорковими утворення ми. Коркова реакція реалізується через активацію норадре нергічних, серотонінергічних, допамінергічних, холінергічних і гістамінергічних нейронів головного мозку і гіпоталамуса [33]. Підкоркова аrousalреакція контролюється центрами стовбура головного мозку, відповідальними за серцевий ритм, артеріальний тиск, респіраторну активність [33]. Показано, що у дітей з груп ризику за СРДС відсутні компенсаторні реакції на виникнення гіпоксії. При цьому підвищення в крові рівня вуглекислого газу і зниження рівня кисню не тільки не стимулює, але ще більше пригнічує дихальний центр. 3. Прогресуюча асфіксія призводить до гіпоксичної коми (втрати свідомості, арефлексії, важких розладів функцій). У дослідах на тваринах було показано швидкий розвиток коматозного стану при досягненні критичного рівня артеріальної оксигенації (близько 10 мм рт. ст.). Експериментально показано розвиток критичної гіпоксії мозку у відповідь на його гіпоперфузію з переходом в гіпо ксичну кому у дітей, згодом загиблих від СРДС]
79. Задишка (диспное). Причини та механізми її розвитку.
Задишка (дисппое) - це відчуття нестачі повітря і пов'язана з цим потреба посилити дихання.
Відчуваючи нестачу повітря, людина не тільки мимовільно, але й свідомо збільшує активність дихальних рухів, прагнучи позбутися цього тяжкого відчуття, наявність якого і є найбільш істотною відмінністю диспное від інших видів порушення регуляції дихання (гіперпное, поліпное та ін.). Тому у людини, що втратила свідомість, задишки не буває.
Задишка виникає у тих випадках, коли впливи, що збуджують вдих, є сильнішими за впливи, що його пригнічують, а також у разі підвищення чутливості дихального центру до чинників, які стимулюють дихання. Найважливішими з цих впливів є такі.
1. Збудження рецепторів, що стимулюють центр вдиху, і які активуються при сильному зменшенні об'єму легеневих альвеол (сильнішому, ніж при максимальному видиху). При патології може виникнути постійна імпульсація від цих рецепторів. Наприклад, при застійних явищах у легенях (недостатність серця, пневмонія) переповнені кров'ю судини, що оточують альвеоли, здавлюють їх, ємність альвеол зменшується, що веде до збудження рецепторів спадіння.
2. Збудження рецепторів інтерстиціальної тканини легень (J-рецепторів). Усі патологічні процеси, що ведуть до застійних явищ у легенях (пневмонія, недостатність серця), можуть викликати тривале збудження J-рецепторів і підвищену стимуляцію дихальних нейронів.
3. Рефлекси з повітроносних шляхів. Можуть викликатися твердими частинками, парами хімічних сполук та іншими чинниками, що потрапили в повітроносні шляхи і подразнюють так звані іритантнірецептори (мають властивості одночасно ме-хано- і хеморецепторів), розташовані в субепітеліальному шарі трахеї, бронхів і бронхіол. Значне збудження іритантних рецепторів спостерігають при бронхітах, бронхопневмонії, бронхіальній астмі і хворобах, при яких у бронхах і альвеолах міститься слиз, ексудат або транссудат).
4. Рефлекси з барорецепторів аорти і сонної пазухи. Ці рефлекси долучаються до патогенезу задишки при крововтраті, шоку, колапсі. За умов зменшення артеріального тиску до рівня 70 мм рт. ст. і нижче, різко зменшується імпульсація від цих рецепторів, яка в нормі гальмує центр вдиху (через активацію центру видиху).
5. Рефлекси з хеморецепторів аорти і сонної пазухи. При зниженні в крові напруги 02, підвищенні напруги С02 або збільшенні концентрації іонів водню відбувається посилене збудження рецепторів, розташованих в аортальному і каротидному тільцях, і, як наслідок, — посилене збудження центру вдиху. Цей механізм відіграє важливу роль у розвитку задишки при ацидозі, недостатності дихання, при анемії і т. д.
6. Безпосередня стимуляція нейронів дихального центру. У довгастому мозку є хеморецептори, вибірково чутливі до вуглекислого газу. Сильне збудження цих рецепторів при гіперкапнії також сприяє розвитку задишки. 7. Рефлекси з дихальних м 'язів. Відчуття нестачі кисню може виникнути при надмірному розтягненні міжреберних м'язів і сильному збудженні рецепторів розтягу, імпульсація від яких надходить у вищі відділи головного мозку. Цей механізм діє під час виконання тяжкої фізичної роботи, що потребує значної роботи інспіра-торних м'язів, при зменшенні еластичності легень, звуженні верхніх дихальних шляхів. 8. Стимуляція дихального центру продуктами власного метаболізму. Ідеться про накопичення вуглекислого газу, кислих продуктів обміну і зниження напруги кисню безпосередньо в нервових центрах унаслідок порушення мозкового кровообігу (спазм або тромбоз судин головного мозку, набряк мозку, колапс). Дихання при задишці, як правило, часте і глибоке. Посилюється як вдих, так і видих, який за умов задишки носить активний характер і відбувається за участю експіраторних м'язів. Однак у деяких випадках може переважати або вдих, або видих, тоді ведуть мову про інспіраторну (посилений вдих) або експіраторну (посилений видих) задишку. Інспіраторну задишку спостерігають, наприклад, у першій стадії асфіксії, при загальному збудженні центральної нервової системи, при фізичному навантаженні у хворих з недостатністю кровообігу, при пневмотораксі. Експіраторна задишка буває рідше і виникає головним чином при бронхіальній астмі, емфіземі, коли при видиху збільшується опір потокові повітря в нижніх дихальних шляхах.
80. Поняття про синдром транзиторного тахіпное новонароджених, його етіопатогенез.
Транзиторне тахіпное новонароджених (ТТН) – це стани у доношених новонароджених або у недоношених дітей у віці старше 34 тижнів внутрішньоутробного розвитку, які виникають після народження у зв’язку із затримкою вивільнення альвеол від внутрішньоутробної легеневої рідини й триває менше 24 годин.
Фактори ризику:
- кесарський розтин (частота – до 20-25%);
-гостра асфіксія;
- надлишкова медикаментозна терапія матері під час пологів;
- пролонговані пологи;
- цукровий діабет у матері.
Збільшення вмісту легеневої рідини та сповільнене її видалення через лімфатичну систему відбувається через різні причини:
- підйом центрального венозного тиску внаслідок надлишкового введення розчинів матері під час пологів;
- пізнє клемування пуповини й потрапляння надлишкової кількості крові від плаценти до плоду;
- при кесарському розтині відсутнє стиснення грудної клітки, що спостерігається при народженні через природні родові шляхи;
- відсутність «катехоламінового сплеску» при проведенні оперативного втручання під час пологів.
Накопичення рідини у периваскулярному просторі та інтерстиціальній тканині легень призводить до стиснення повітроносних шляхів, що обумовлює розвиток емфіземи. Можливий також й інший механізм розвитку емфіземи – при частковій обструкції дихальних шляхів повітря за типом «клапанного» або «поплавкового» механізму під час вдиху потрапляє в ацинуси, а під час видоху не може вийти звідти через зменшення просвіту бронхіол.
На рентгенограмі органів грудної клітини визначається посилений легеневий малюнок, підвищена прозорість периферичних ділянок легень, сплощений купол діафрагми. У ряде випадків у реберно-плевральних синусах може відзначатися накопичення рідини. Необхідно виключити інші причини дистресу, в тому числі пневмонію, вроджені вади серця, синдром поліцитемії, спадкові хвороби обміну речовин.
При ТТН виявляються помірні прояви гіпоксемії, метаболічного ацидозу. Через тахіпное зазвичай визначається низький рівень РаСО2.
Необхідно підтримувати оптимальний температурний режим. Ентеральне харчування починають при зменшенні задишки. Інфузійна терапія призначається, виходячи з мінімальних енергетичних та водно-електролітних потреб. Для підтримання SaО2 у нормальних межах проводять інгаляцію 40-50% повітряно-кисневої суміші. Штучну вентиляцію легень застосовують в дуже рідкісних випадках.
81. Поняття про синдром раптової смерті немовлят, фактори ризику, гіпотези патогенезу.
Синдром раптової дитячої смерті (СРДС) — несподівана ненасильницька смерть здорової дитини першого року життя, при якій відсутні адекватні для пояснення причини смерті дані анамнезу та патоморфологічного дослідження.
Виділяють дві основні групи факторів ризику СРДС: соціальнодемографічні та перинатальні. Соціально-демографічні фактори ризику асоціюються з низьким соціальним і матеріальним статусом сім'ї, недостатнім освітнім рівнем батьків, неповною сім'єю, поганими мате ріальними та побутовими умовами. Серед антенатальних факторів ризику мають значення повторні пологи (другі і більше), невеликий часовий інтервал між даними і попередніми пологами (менше 14 міс.), юний вік матері (молодше 17 років), відсутність адекватного спостереження за вагітною, шкідливі звички матері (алкоголь, вживання наркотиків, паління), затрим ка внутрішньоутробного розвитку плода.
До «побічних» факторів належать:
Сон на животі;
Низька вага при народженні;
Підвищена або ж знижена кімнатна темпера
82. Етіопатогенез гострого респіраторного дистрес-синдрому новонароджених. Принципи лікування та попередження.
Гострий респіраторний дистрес-синдром (ГРДС), також респіраторний дистрес-синдром дорослих (РДСД) — загрозливе для життя запальне ураження легень, для якого характерні дифузна інфільтрація[en] та тяжка гіпоксемія. Виникає внаслідок багатьох причин, які безпосередньо чи опосередковано уражають легені. ГРДС часто призводить до смерті, потребує проведення інтенсивної терапії[en] та штучної вентиляції легень[en].
Етіологія
ГРДС настає внаслідок прямого або непрямого ушкодження легень. Непряме ушкодження легень виникає внаслідок системної запальної реакції при позалегеневих захворюваннях. До найчастіших причин відносять сепсис та/або пневмонію (зокрема аспіраційну[en]), тяжкі травми. Інші причини представлено в таблиці[7].
|
Прямепошкодженнялегень |
Непрямепошкодженнялегень |
|
Аспірація |
Сепсис |
|
Пневмонія |
Важка травма |
|
Дифузна альвеолярна кровотеча |
Пересадка кісткового мозку |
|
Жирова емболія |
|
|
Пересадка легень |
Кардіопульмональний шунт |
|
Передозування ліків (аспірин, кокаїн, опіати, фенотіазини[en], трициклічні антидепресанти) |
|
|
Контузія легкого |
Масивне переливаннякрові |
|
Вдихання токсичного газу |
Неврогенний набряк легеніввнаслідок інсульту, судом, травмиголови |
|
Рентгеноконтрастніпрепарати (рідко) |
Патогенез
В основі ГРДС лежить дифузне запалення легень. У його перебігу виділяють 3 фази: ексудативну, проліферативну та фібротивну[6].
Під час ексудативної фази у відповідь на запалення виділяються цитокіни та інші прозапальні речовини, які активують альвеолярні макрофаги і циркулюючі нейтрофіли. Самі ж активовані нейтрофіли прикріплюються до ендотелію легеневих капілярів і вивільняють вміст своїх цитоплазматичних гранул[en] (протеази і токсичні метаболіти кисню)[8]. Це призводить до пошкодження ендотелію капілярів та епітелію альвеол, порушуючи альвеолярно-капілярний бар'єр. Внаслідок цього ексудат проходить у легеневу паренхіму і альвеолярний повітряний простір. Порушується газообмін і настає гіпоксія[9]. Також можливе пошкодження альвеолоцитів[en] II типу, які відповідають за утворення сурфактанту. Це призводить до спадання альвеол, зниження розтяжності легень і внутрішньолегеневого шунтування. Крім того, розвивається легенева гіпертензія внаслідок внутрішньосудинної обтурації тромбами, спазму легеневих судин через гіпоксію й дію деяких запальних медіаторів (тромбоксан[en], лейкотрієни та ендотелін[en])[10].
Під час проліферативної фази в більшості пацієнтів відбувається відновлення легень: виводиться ексудат, замість нейтрофільної інфільтрації настає лімфоцитарна. Проліферують альвеолоцити II типу, що утворюють новий сурфактант і диференціюються в альвеолоцити I типу. Але попри такі поліпшення, у багатьох хворих зберігається задишка, тахіпное, гіпоксемія[6]. У деяких пацієнтів процес переходить у фібротичну фазу. Накопичений в легенях фібрин зазнає ремоделювання й може викликати фіброз[11].
Лікування
Лікування передусім спрямовують на усунення захворювання, що призвело до ГРДС. Якщо це неможливо (наприклад, після масивних переливань крові, аортокоронарного шунтування і т. д.), то обмежуються підтримувальною терапією.
Діагностика(попередження)
Виявляють прогресуючу гіпоксемію (SpO2 нижче 90 %), яка часто рефрактерна до інгаляцій кисню[3]. Досліджуючи гази артеріальної крові[en] на початкових стадіях ГРДС виявляють низький PaO2, нормальний або низький РаСО2 і підвищення рН (алкалоз). Надалі РаСО2 наростає і алкалоз змінюється ацидозом[9].
На рентгенографії легень видно двобічні дифузні інфільтрати, іноді — плевральний випіт. Такі ознаки неспецифічні й також характерні для кардіогенного набряку легень, що ускладнює диференціальну діагностику[en][13][14]. Комп'ютерна томографія показує негомогенну інфільтрацію легень у деяких відділах (у задньонижних відділах у лежачих хворих)[15]. Це пояснюється тим, що розподіл набряку легень залежить від сили тяжіння, а також тиском з боку розташованих вище набряклих відділів легень[16].
Бронхоальвеолярний лаваж — найнадійніший метод діагностики ГРДС. Під час нього вводять гнучкий фібробронхоскоп в один з уражених сегментів легенів. Потім промивають легеневий сегмент ізотонічним розчином і аналізують склад промивної рідини[3]. У хворих з ГРДС виявляють нейтрофіли, що становлять 60-80 % всіх клітин промивної рідини (в нормі < 5 %)
83. Порушення травлення в порожнині рота. Етіологія, патогенез карієсу та пародонтозу. Причини, механізми та наслідки порушень слиновиділення.
Патології порожнини рота
Причини розладу акту жування
У порожнині рота їжа подрібнюється зубами і змочується слиною. Розлад акту жування виникає внаслідок ушкодження або відсутності зубів, порушення функції жувальних м'язів чи скроне-нижньощелепних суглобів, захворювання слизової оболонки порожнини рота. Причиною ушкодження або втрати зубів здебільшого є карієс і періодонтит.
Карієс зубів
Карієс зубів — патологічний процес, що характеризується прогресуючою деструкцією твердих тканин зуба (емалі й дентину) з утворенням дефекту у вигляді порожнини. Поширеність карієсу зубів у деяких районах земної кулі досягає 100%.
Періодонтит
Періодонтит (англ. Periodontitis) спостерігається у всьому світі у 30-50% людей у віці після 30 років. Це запально-дистрофічний процес, що уражає тканини періодонта (тобто тканини, що оточують корінь зуба — періодонт, кісткова тканина зубної комірки, ясна, окістя). Проявом пародонтиту є резорбція зубних комірок, гноєтеча з ясенних закутків, розхитування і випадання зубів.
Порушення функції слинних залоз також спричинює розлади обробки їжі в порожнині рота. Підвищене слиновиділення — гіперсалівація — виникає внаслідок запалення слизової оболонки порожнини рота (стоматит, гінгівіт). На процес слиноутворення рефлекторно можуть впливати зуби, уражені патологічним процесом. Гіперсалівація може супроводжувати захворювання органів травлення, а також блювання, вагітність, може виникати під впливом фосфорорганічних та бойових отруйних речовин. Зниження секреції слини — гіпосалівація — виникає під час інфекційних захворювань і гарячкових станів, при зневодненні, під впливом атропіну, а також у зв'язку із запальним процесом у слинних залозах. Гіпосалівація утруднює акт жування і ковтання, сприяє виникненню запальних процесів у слизовій оболонці порожнини рота і проникненню інфекції у слинні залози, а також розвитку карієсу зубів. Недостатня попередня обробка їжі може виникати внаслідок розладів акту жування. Причиною розладу жування, окрім травм і звапальних процесів, може бути порушення іннервації жувальних м'язів. У порожнині рота знаходиться близько 30 видів мікроорганізмів, серед яких немало патогенних. Мікрофлора, що розвивається на фоні карієсу зубів, пародонтиту, внаслідок ангін, хронічного тонзиліту, може зумовлювати сенсибілізацію організму і навіть спричиняти оральний сепсис.
Етіологія- Сучасна концепція етіології карієсу.
Грунтуючись на історичних теоріях, в даний час досягнуті значні успіхи у вивченні етіології і патогенезу карієсу зубів.
Загальновизнаним механізмом виникнення карієсу є прогресуюча демінералізація твердих тканин зубів під дією органічних кислот, утворення яких пов'язане з діяльністю мікроорганізмів.
У виникненні каріозного процесу беруть участь безліч етіологічних чинників, що дозволяє рахувати карієс поліетіологичним захворюванням. Основними етіологічними чинниками є:
1) мікрофлора порожнини рота;
2) характер і режим харчування, вміст фтору у воді;
3) кількість і якість слиновиділення;
4) загальний стан організму;
5) екстремальні дії на організм.
Всі вищеперелічені чинники були названі карієсогенними і підрозділені на загальні і місцеві, такі, що відіграють важливу роль у виникненні карієсу.
Патогенез
1) анатомічна будова зуба і взаємовідношення його з навколишніми тканинами;
2) структура поверхні зуба;
3) харчовий раціон і інтенсивність жування;
4) слина і ясенна рідина;
5) гігієна порожнини рота;
6) наявність пломб і протезів в порожнині рота;
7)зубощелепні аномалії.
Этиология пародонтита. Ведущим этиологическим фактором пародонтита, по данным отечественных и зарубежных исследователей, является микрофлора зубной бляшки, образующейся на пелликуле зуба в области
зубодесневой борозды. Патогенное влияние микрофлоры может быть связано с изменением её состава при избыточном накоплении в зубном налёте. В этих случаях появляются преимущественно грамотрицательные микроорганизмы, фузобактерии, спирохеты. В последние годы в зубном налете, вызывающем воспаление и деструкцию тканей пародонта, отмечают роль так называемых ассоциаций потенциально агрессивной микрофлоры: Actinobacillus, Actinomycetemcomitans, Porphyromonas gingivalis, Bacteroides forsytus, Spirochetе, Prevotella intermedia, Campilobacter rectus, Eubacteriuv nodatum, Treponema denticola, Streptococcus intermedius, Peptostreptococcus micros, Fusobacterium nucleayum, Eikenella corrodens.
Выделяют ряд местных и общих факторов риска, способствующих возникновению пародонтита. Факторы, вызывающие перегрузку пародонта: патология прикуса (скученность зубов), супраконтакты, травматические «узлы», парафункциональные привычки (сжатие зубов, бруксизм), дефекты протезирования и пломбирования. Факторы, вызывающие ишемию тканей пародонта, – короткие уздечки языка и губ, нарушение прикрепления уздечек языка, губ и тяжей, мелкое преддверие рта. Плохая гигиена полости рта, придесневые кариозные полости предрасполагают к развитию пародонтита. Врожденные особенности строения пародонта: тонкая, малокератинизированная десна, недостаточная толщина альвеолярной кости, выпуклый контур зубной дуги, часто сочетающийся с выпуклостью корней.
Общие заболевания, связанные с нарушением процессов адаптации: хронические эмоциональные стрессы, эндокринные заболевания, мочекаменная болезнь, язвенная болезнь ЖКТ, системный остеопороз и др. соматическая патология.
Все перечисленные факторы, нарушая защитную систему пародонта, создают предпосылки к реализации патогенного влияния микрофлоры на ткани пародонта и в первую очередь – на зубодесневое прикрепление, воспаление и деструкция которого является началом пародонтита.
Патогенез. Особенности влияния зубной бляшки на развитие пародонтита:
- активное воздействие протеолитических ферментов, которые, действуя на межклеточные связи эпителия прикрепления, приводят к повышению его проницаемости;
- кроме этого, действуя на органическую субстанцию эпителиального прикрепления, ферменты изменяют коллоидное состояние и способствуют нарушению связи эпителия с эмалью зуба;
- образуемые анаэробными бактериями эндотоксины повреждают клетки, соединительнотканные образования и основное вещество. Они могут активировать систему комплемента, кининов и других медиаторов воспаления, вызывая ответные иммунные реакции – гуморальные и клеточные, способствовать развитию воспаления мягких тканей с последующей деструкцией костной ткани альвеолы;
- секретируемые в процессе воспаления биологически активные вещества (гистамин, серотонин) воздействуют на клеточные мембраны сосудов – прекапилляров и капилляров. Биологически активные вещества активизируют выход форменных элементов крови, активизируют тучные и плазматические клетки, лимфоциты;
- патогенная микрофлора, обладая антигенными свойствами и оказывая сенсибилизирующее действие, приводит к усилению альтерации и образованию аутоантигенов, которые вызывают лизис круговой связки зуба, костной ткани. При этом освобождаются новые тканевые антигены, которые усугубляют течение пародонтита.
Здесь тоже много полезного и в конце ответ на причины и последствия и механизм
Порушення травлення в порожнині рота. Етіологія, патогенез карієсу, пародонтозу. Причини, механізми, наслідки порушень слиновиділення
Порушення травлення в порожнині рота: 1)порушення моторної ф-ї (порушення жування внаслідок карієсу, пародонтиту, міохиту, невриту, бульбарного паралічу, ураж.суглобів, травми щелеп, ураж.язика тощо) 2)секреторної ф-ї (гіпо та гіперсалівація). Карієс зубів це патологічний процес, що виявляється демінералізацією та прогресуючим руйнуванням тканин зуба з утворенням дефектів та порожнин. Етіологія: мікроорганізми (стрептокои, лактобакитерії), надмірне вживання вуглеводів які легко ферментуються бактеріями, недостатність в харчуванні вітам.гр.В Са Р мікроелементів, генетичний ф-р, пор.захисної ф-ї слини, гипокальци(фосфор)емічні стани. Патогенез: утв.зубної бляшки - скопичення м/о та продуктів їх метаболізму, в т.ч. карієсогенних ф-рів (органічні к-ти розчиняють оксиапатит емалі та дентину, р-ни-хелатори пептиди та АК утв.комплекси з Са вивільнюючи його з оксиапатитів, протеолітичні ферменти). Також зниження захисту слини, багато вуглеводів мікрофлора. Також роль фосфорно-кальцієвого обміну. Пародонтит це дистрофічно- запально аутоімунне ураження пародонта з руйнуванням зубодесняного сполучення та прогресуючою деструкцією альвеолярних відростків, що веде до випадіння зубів. Етіологія і патогенез: мікрофлора (ферменти та БАР зубний наліт, камінь порушення трофіки тканин зуба), аліментарні пор. (дефіцит віт.С,Р,Е), нейрогенні ф-ри (стрес, пор.нервової трофіки), ендокринні пор. (гіпогонадизм, гіпотиреоз, гіпоінсулінізм, гіперпаратиреоз) та аномалії прикусу (перевантаження). Гіперсалівація. (5-20л) Етіологія і патогенез: рефлекторний мех (збудж.рец-рів роту, стравоходу, шлунку, запальні процеси в роті, бормашина), збудж.центру слиновиділення (бульбарний параліч), подраз.вегетат.нервів (парасимп багато рідкої, симп -мало густої слини), вплив М-холіноміметиків, вплив гумор.ф-рів (в основі паралітичної гіперсалівації, через добу після денервації слинних з-з збільшується вплив АХ,гістаміну, але через 40 днів салівація зникає). Наслідки: гіперосмолярна гіпогідрія, нейтралізація шлункового соку. Гіпосалівація. Причини:центральне гальмування (страх, біль), М-холінолітики, запалення пухлини слинних з-з, закупорка проток каменями, зневоднення. Наслідки: порушення жування, ковтання, травматизація слизової і стоматит, мало лізоциму мікрофлора, пор.трофіки зубів).
84.Причини порушення функцій стравоходу. Дивертікули стравоходу. Гастроезофагальн рефлексна хвороба.
Нарушения функций пищевода
Ахалазия пищевода (греч. «а» - отсутствие, «халазис» - расслабление) – заболевание, основной причиной которого является дегенерация нервных ганглиев в стенке пищевода.
Увеличение внутрипищеводного давления наблюдается при ахалазии пищевода; диффузном спазме мышц пищевода; «гипертонусе» нижнего пищеводного сфинктера, при системной склеродермии, сахарном диабете, интоксикациях алкоголем и его суррогатами, а также при психических расстройствах (истерии).
Снижение тонуса НПС является основным звеном патогенеза гастроэзофагеальной рефлюксной болезни (ГЭРБ).
Гастроэзофагеальная рефлюксная болезнь характеризуется частыми (более 50 раз в течение часа) эпизодами расслабления НПС, попаданием кислого содержимого желудка в пищевод и повреждением вследствие этого слизистой оболочки пищевода. Причины, приводящие к возникновению ГЭРБ, многочисленные:
Растяжение кардиального отдела желудка
Значительное увеличение внутрибрюшного давления
Наличие грыжи пищеводного отверстия диафрагмы
Анатомические нарушения строения НПС
Угнетение нормальной перистальтической активности пищевода
Гипосаливация, снижение рН слюны
Активация блуждающего нерва
Функциональные причины: употребление кофе, алкоголя; прием некоторых лекарственных препаратов (антагонистов кальциевых каналов, нитратов, β-адреноблокаторов); увеличение содержания прогестерона в крови или прием синтетических гестагенов; повышение образования вазоинтестинального пептида, оксида азота и гистидинизолейцина, расслабляющих НПС.
Дивертикул стравоходу - мішкоподібне випинання стінки стравоходу, яке сполучається з його просвітом.
Гастроезофагеальна рефлюксна хвороба (ГЕРХ) – хронічне рецидивуюче захворювання, що проявляється характерними симптомами, які турбують хворого, і/або запаленням дистальних відділів стравоходу (рефлюкс-езофагітом) внаслідок гастроезофагеального рефлюксу – регулярного закиду в стравохід шлункового і/або дуаденального вмісту.
85. Порушення секреторної функцій шлунка. Патологічна шлункова секреція, види; причини та механізми розвитку. Роль нервових та гуморальних механізмів у порушенні секреції.
Экзогенные (чаще.первичные) функциональные расстройства желудка встречаются при алиментарных нарушениях: нерегулярный прием пищи, быстрая смена режима питания, быстрая еда и еда всухомятку, переедание, злоупотребление углеводами, свиным и бараньим жиром, грубой растительной клетчаткой, грибами, копченостями, специями, недостаточное количество белка, витаминов, минеральных солей, микроэлементов.
Эндогенные (чаще вторичные) функциональные расстройства желудка возникают при заболеваниях других органов и систем: нервной, эндокринной, пищеварительной, сердечно-сосудистой, дыхательной, мочевыделительной, кроветворной.
Особенно часто причиной эндогенных функциональных расстройств желудка являются болезни органов пищеварения. Отмечено, что при заболеваниях кишечника (хронический энтероколит, дизентерия) желудочная секреция вначале усиливается, а затем резко снижается, сопровождаясь угнетением двигательной функции желудка. При хроническом холецистите снижаются как секреторная, так и двигательная функции желудка вплоть до ахлоргидрии и гипотонии. Разнонаправленные изменения секреторной деятельности желудка наблюдаются при остром вирусном гепатите.
При различных патологических процессах в желудке натощак отделяется качественно различный желудочный сок. В частности, при гнойных, септических процессах, силикозе в желудке обнаруживается большое количество секрета с низким уровнем соляной кислоты и малой переваривающей силой сока, а иногда и полное отсутствие соляной кислоты. У больных сепсисом за 3—4 часа выделяется до 400—600 мл желудочного сока, не содержащего соляной кислоты.
Фази шлункової секреції — це фази активації утворення виділення шлункового соку, які зумовлені різними нервові гуморальними регуляторними механізмами. У мозкову (складно-рефлекторну) фазу відбувається активація шлункового соковиділення на вигляд, запах, підготовку їжі для споживання через рецептори зору, слуху, (умовнорефлекторне збудження) та при надходженні їжі в ротову порожнину і збудженні тим самим рецепторів роту, язика, піднебіння, глотки (безумовнорефлекторна секреція шлункова (нервово-гуморальна) фаза відбувається при механічному і хімічному подразненні рецепторів слизової шлунка їжею, а також під впливом гуморальних факторів (гістаміну, гастрину тощо); кишкова фаза настає при надходження до кишківника шлункового вмісту, що викликає виділення ендокриноцитами слизової оболонки кишківника гормонів, зокрема ентерогастрину (головний найпотужніший гуморальний фактор), які через кров стимулюють виділена шлункового соку.
86.Виразкова хвороба. Етіологія та патогенез. Фактори ушкодження, причини зниження захисних властивостей та регенераторної здатності шлунку та 12-палої кишки. Патогенез виразок при стресі.
Виразкова хвороба - це хронічне захворювання шлунка і дванадцятипалої кишки, яке протікає циклічно, характеризується так званою сезонністю, утворенням виразок в період загострення. Виникає виразкова хвороба як наслідок розладу гуморальної і ендокринної регуляції секреторних і моторних процесів, а також порушень захисних механізмів слизової оболонки гастродуоденальноі зони.
Етіологія. Виразкова хвороба- це поліетіологічне захворювання. Тепер описано понад 40 факторів ризику виникнення виразкової хвороби. Їх можна поділити на дві валикі групи: основні фактори які обумовлюють розвиток захворювання і фактори, які сприяють цьому патогенна роль яких визначається при наявності основних факторів.
За данними В.І.Маколкіна (1987) до основних факторів ризику виникнення виразкової хвороби відносяться:
- нервово-психічні перенапруження, гострі й хронічні психічні травми, баротравми, закриті черепно-мозкові пошкодження, розлади діяльності ендокринних залоз;
хронічні захворювання шлунка і дванадцятипалої кишки, зокрема гастрити, гастродуоденіти, бульбіти, дуоденіти, які протікають з ушкодженням слизової оболонки і порушеннями шлункової секреції і моторики;
порушення ритму й характеру харчування.
До факторів, які сприяють розвитку виразкової хвороби відносяться такі, як:
- конституційні й спадкові особливості, в тому числі збільшення, кількості обкладальних клітин (тих, що виробляють соляну кислоту), група крові ( ті, що мають 0 (І) групу крові хворіють частіше, ніж інші);
- вплив зовнішнього середовища (ритмічні зміни вологості, атмосферного тиску, температура);
- зловживанням курінням та алкоголем;
- супутні захворювання (гепатити, цирози печінки).
Починаючи з 1983 року посилено вивчається й інша точка зору на етіологію виразкової хвороби. Мова йде про роль мікробного інфікування слизової оболонки антрального відділу шлунка.
Цей мікроорганізм виявляють у 75-процентів хворих на виразкову хворобу дванадцятипалої кишки. Він є анаеробом, розташовується під шаром слизу на поверхні шлункового чи дуоденального епітелію. Інфікування антрального відділу шлунка зростає в разі поверхневого гастриту незалежно від того чи є гастрит самостійним захворюванням чи супутником виразкової хвороби.
Патогенез. Механізм виникнення виразкової хвороби вивчений ще недостатньо. Припускають, що під впливом вказаних вище етіологічних чинників порушується функціональний стан кори великих півкуль і гіпоталамо- гіпофізарноі системи, підвищується активність парасимпатичної нервової системи (таблиця 1). Внаслідок переважання парасимпатичної нервової системи над симпатичною посилюється секреція шлункового секрету та моторна фрункція шлунка. Все це в поєднанні з спадково-конституційнимими особливостями (збільшення кількості обкладальних клітин, які виробляють соляну кислоту) підвищує активність кислотно-пептичного чинника. Доречі, цьому сприяє також підвищення рівня гастрину (як наслідок підвищеної секреції наднирниковими залозами кортизону) бо при цьому захворюванні посилюється функція передньої долі гіпофіза. До того ж, зміна функціональної активності наднирникових залоз веде до пониження опірності слизової оболонки щодо дії кислотно- пептичного чинника. Поряд з цим відбувається зниження регенераторної здатності слизової оболонки, падає захисна функція слизистого бар’єру внаслідок зменшення секреції слизу. Порушення моторної функції шлунка, зокрема прискорення його спорожнення. За сучасними уявленнями обсіменіння слизової оболонки гастродуоденальної зониHeliobacter pylori (H. pylori) є важливим фактором рецидиву виразки.
Що ж торкається виразкової хвороби шлунка, то при цьому спов1льнюється евакуація шлункового вмісту в дванадцятипалу кишку. З другого боку при цьому захисний компенсаторний механізм ретропульсаціі /періодичне поступання лужного вмісту дванадцятипалої кишки в шлунок, внаслідок чого нейтралізується кислотнопептична агресія в антральному відділі шлунка/ поступово переходить в патологічний гастродуодзнальний рефлюкс.
При виразковій хворобі дванадцятипалої кишки найбільше значення має підвищення активності кислотно-пептичного фактору в поєднанні з прискореним спорожненням шлунка. В разі ж виразки шлунка провідне значення має ураження слизової оболонки шлунка внаслідок зниження її регенераторної здатності. До того ж при виразковій хворобі шлунка має місце зниження ефективності слизистого /мукоциліарного/ бар'єру в поєднанні із сповільненням авакуації шлунка.
Патогенез стрессовых язв
Установлено, что желудок является одной из главных мишеней органных повреждений у больных, находящихся в критическом состоянии, в связи с тем что его слизистая оболочка постоянно контактирует с внутрипросветными эндогенными повреждающими субстанциями, к которым относятся соляная кислота, пепсин, а также забрасываемые в желудок желчь и панкреатические ферменты. Важная роль в повреждении слизистой оболочки желудка (СОЖ) принадлежит также гематогенным эндогенным повреждающим факторам. К ним относятся: провоспалительные и вазоактивные медиаторы, продукты нарушенных метаболических процессов в организме (мочевина, мочевая кислота и др.) и ряд экзогенных повреждающих субстанций (вирусы, бактерии и их токсины, лекарства, химические вещества, соли тяжелых металлов и др.), поступающих в организм, минуя желудочно-кишечный тракт. Под влиянием экзогенных и эндогенных ксенобиотиков в СОЖ нарушаются метаболические процессы, что приводит к появлению новых биологических субстратов, оказывающих деструктивный эффект.
Основные механизмы действия повреждающих СОЖ агентов:
падение трансмембранного потенциала поверхностного эпителия, способствующего обратной диффузии водородных ионов в СОЖ;
деградация пристеночной слизи и повреждение поверхностного эпителия СОЖ;
индукция синтеза провоспалительных субстанций в СОЖ;
блокада продукции противовоспалительных тканевых простагландинов и АТФ;
активация тканевых фосфолипаз и перекисного окисления липидов в эпителии СОЖ;
нарушение кровотока и сосудистой проницаемости в СОЖ.
87. Системні та місцеві фактори ульцерогенезу. Захисні механізми при виразковій хворобі. Ваги Шія.
Фактори ульцерогенезу зв’язують з тим, що НР може виділяти протеазу, фосфоліпазу і цитотоксини, порушувати цілісність поверхневого епітелію і руйнувати слизовий бар’єр, создаючи умови для протеолізу стінки шлунка.
Місцеві фактори ульцерогенезу:
12-палої кишки:Helicobacter pylori-92% ; HПВС-5%; інші-3%
Шлунка: Helicobacter pylori-70%; НПВС-25%; інші-5%
Затисну роль відіграють слизовий бар’єр, який перешкоджає зворотній дифузії іонів водню та їхній ушкоджувальній дії, алекватне кровопостачання і високий регенеративний потенціал слизової оболонки. Таку саму роль відіграють нейтралізація кислого секрету гідрокарбонатами, які разом із слизом виробляються клітинами поверхневого епітелію, ростові і трофічні фактори, що сприяють регенерації, простогландини та інші гормони. Гідрокарбонати виробляються і парієтальними клітинами, які секретують Cl(-) i H(+) з апікальної поверхні у шлунок, HCO3(-) i K(+) з базальної поверхні в підслизовий прошарок.
88. Пептична виразка шлунку – причини та механізми виникнення. Патогенетичне обґрунтування напрямків корекції.
Головною причиною хвороби (понад 60% виразок шлунку і 90% виразок дванадцятипалої кишки) є хронічна інфекція Helicobacter pylori, спиралеподібна бактерія, яка втім не є спірохетою, а протеобактерією, що колонізує (тобто надовго поселяється, коли потрапляє до тіла) антральну слизову оболонку. Імунна система не в змозі знищити інфекцію, не зважаючи на появу антитіл. Тому бактерія може вчинити хронічний активний гастрит (гастрит типу B) або дуоденіт, приводячи до дефектів у регулюванні виробництва гормону гастрину цією частиною шлунку або дванадцятипалої кишки, що зумовлює збільшення секреції цього гормону. Гастрин, у свою чергу, стимулює виробництво шлункової кислоти тім'яними клітинами. Кислота роз'їдає слизову оболонку і спричинює пептичну виразку.
Інша важлива причина пептичної виразки — використання НПЗП. Слизова оболонка шлунку захищає себе від власної соляної кислоти за допомогою шару слизу, секрецію якого стимулюють деякі простагландини. НПЗП блокують функцію циклооксигенази-1 (ген cox-1), яка істотна для виробництва простагландинів. Новіші NSAID (целекоксиб, рофекоксиб) інгібують тільки cox-2, яка менш істотна для функції шлунковій слизової оболонки, і зменшують ризик виникнення пов'язаної з НПЗП пептичної виразки приблизно вдвічі.
Глюкокортикоїди приводять до атрофії всіх епітеліальних тканин. Проте, їх роль у виникненні пептичної виразки невелика.
Стрес також практично не впливає на розвиток пептичних виразок. Серйозні травми, проте, інколи приводять до «стресових виразок» у багатьох пацієнтах, які знаходяться на штучній вентиляції легень.
Куріння також має роль у виникненні пептичних виразок. Воно приводить до атеросклерозу і спазму судин, зумовлює судинну недостатність, сповільнюючи швидкість репаративних процесів і підсилюючи секрецію шлунку, таким чином просуваючи розвиток виразок через ішемію судин і слизової.
Спадковість інколи відіграє роль у розвитку виразок дванадцятипалої кишки, особливо, за спадкування групи крові O(I). Можливо, що спадковість не має значення у випадку виразок шлунку.
Гастрінома (пухлина острівкового апарату підшлункової залози, також відома як синдром Золлінгера — Еллісона), та рідкісні пухлини, що виділяють гастрін, спричинюють багаторазові та тяжкі в лікування пептичні виразки, що складають близько 4% виразок шлунку та незначну кількість виразок дванадцятипалої кишки
89. Роль хелікобактер пілори у виникненні виразок шлунку та 12-палої кишки. Стадії та механізми ульцерогенезу. Патогенетичне обґрунтування діагностики та напрямків лікування.
Опинившись в шлунку Helicobacter, починає активно розмножуватися .Він виробляє особливі ферменти (уреазу, протеази), які пошкоджують захисний шар слизової (внутрішньої) оболонки шлунку і 12-палої кишки, порушує функції клітин, вироблення слизу , обмінні процеси і викликає утворення виразок.
Лікування ВХ шлунка і дванадцятипалої кишки включає проведення медикаментозної та немедикаментозної схеми терапії. До медикаментозно- го варіанту належить призначення антибактеріальних, антихелікобактерних препаратів, гастропротекторов, прокінетиків, засобів, що містять вісмут і анти- секреторних препаратів.
Найбільш важливими завданнями при лікуванні виразкової хвороби є усу- нення симптомів загострення ВХ (болю та диспептичних розладів), досягнення в найкоротші терміни загоєння виразкового дефекту та попередження рецидивів захворювання. У періоди ремісій основне завдання – запобігти повторним заго- стрення, відновити захисні сили організму і здатність до природної саморегуляції.
Сучасні методи лікування виразки шлунка побудовані так, що в результаті успішної ерадикації бактерії відразу настає регенерація тканин і загоєння вираз- ки. Немає необхідності застосовувати додаткові антисекреторні засоби. Лікувальне харчу- вання при ВХ полягає в наступному:
- раціон повинен мати підвищену поживну цінність і включати достатню кількість жирів, білків, вуглеводів, мінеральних солей і вітамінів (обов’язково А, С і вітаміни групи В);
- їжа не повинна подразнювати слизову оболонку шлунку і дванадцятипалої
кишки;
- харчування має бути частим – приблизно кожні 3-4 год.;
- забороняється вживати дуже гарячу або холодну їжу, вона має бути
кімнатної температури;
- продукти, що входять в раціон, не повинні мати сильної сокогонної дії - необхідно скоротити щоденне вживання кухонної солі до 10-12 г.
При ВХ шлунку і дванадцятипалої кишки необхідно виключити з раціону
м’ясні і рибні блюда (жирні сорти м’яса і риби, смажене м’ясо і рибу), міцні м’ясні, овочеві та грибні бульйони, солоні і гострі блюда, ковбаси, консерви, хліб і хлібобулочні вироби (здобне тісто, житній хліб), алкогольні напої, морозиво, міцний чай і каву, солоне і копчене м’ясо і рибу, перець, гірчицю, цибулю, часник. Із медикаментозних засобів, крім патогенетичних і симптоматичних, використовують неспецифічну фармакотерапію (стимулятори імуногенезу, вітаміни, ферментні препарати і ін.). Фармакотерапевтичні засоби сприяютьліквідації залишкових патологічних змін в органах і системах, нормалізують реактивність організму і підвищують фізичну і розумову працездатність. Суттєву роль, при більшості захворювань, на етапах реабілітації відіграє психотерапія, оскільки у значної частини хворих виникають тривожні стани зумовлені невро- тичною фіксацією на своїй неповноцінності, а це сповільнює процеси видужання і адаптації.Так, для стимуляції секреторної діяльності, зменшення швидкості евакуації шлункового вмісту воду п’ють за 10-15 хв. до їжі, а при підвищеній секреції і кислотності шлункового соку – за 1,5 год. Проходячи далі травним шляхом мінеральна вода позитивно впливає на тонкий і товстий кишковик, секреторну функцію підшлункової залози, жовчовивідну функцію печінки.
90. Порушення травлення в кишках, етіологія, патогенез. Синдром мальдигестії: визначення поняття, прояви.
Травлення в кишечнику пов'язане з функцією підшлункової залози, печінки і шлунку.
Ферменти підшлункової залози /протеаза, ліпаза, карбогідраза/ розщеплюють білки, жири, вуглеводи, корм. При посиленому надходженні в кишечник соляної кислоти з кормом збільшується утворення слизового секретииа, який викликає сильну секрецію панкреатичного соку. Особливо це виражено у свиней і жуйних.
Знижується надходження соку підшлункової залози при низькому вмісту соляної кислоти в шлунку, при порушенні прохідності в протоці залози від стискання її пухлинами, рубцями, при запаленні - панкреатиті.
Причини панкреатиту: отруєння, гельмінти і т.д.
Нестача панкреатичного соку знижує розщеплення білків, жирів і приводить до гниття в кишечнику.
Порушення секреції жовчі проявляється нестачею або відсутністю її. Нестача виділення жовчі - гіпохолія. Виникає при закупорках жовчної протоки, каменями, паразитами, пухлинами, при зниженні скоротливої здатності жовчного міхура. Жовч нагромаджується в печінці, всмоктується в кров і виникає жовтяниця. Нестача жовчі в кишечнику знижує емульгування жирів, зменшує дію на них ферментів. Порушення розщеплення і всмоктування жирійв приводить до видалення їх з калом, недостатнього засвоєння жиророзчинних вітамінів /А, Д, Е, К/ і ненасичених жирних кислот. В той час нерозщеплений жир обволікає білок корму і перешкоджає дії на нього ферменту трипсину.
Відсутність надходження жовчі в кишечник /ахолія/ створює сприятливі умови для розвитку бактерій, які викликають в кишечнику процеси гниття і бродіння, ослаблює перистальтику і створює умови для нагромадження вмістимого в тонкому кишечнику /хімостаз/ і газів /метеоризм/.
Порушення секреції кишкового соку.
В кишковому соці містяться ферменти: протеаза, карбогідраза, ліпаза, лактаза та ін. Кишкові залози секретують постійно. Соковиділення посилюється під впливом корму, соку підшлункової залози, при механічному, хімічному, термічному подразнені слизової оболонки кишечника. При запаленні кишечника соковиділення посилюється з утворенням слизу.
В переробці кормів в шлунково-кишковому тракті тварин важливу роль, наряду з порожнинним травленням, відіграє пристінкове /контактне/ травлення. Порожнинне травлення проходить в травному каналі і полягає в розщепленні складних структур їжі, завершальний етап розщеплення їжі здійснюється на поверхні кишкових клітин /ентероцитів/. Цей стан травлення називаємся мембранним.
В основі порушення мембранного травлення лежать слідуючі чинники: порушення структури ворсинок, кишкових клітин, розлади моторики. Так, при деяких інфекційних хворобах, після інтенсивного застосування антибіотиків, кількість ворсинок зменшується від атрофії, що приводить до зменшення поверхні травлення, кишковий епітелій уражається і при променевій хворобі. Порушення всмоктування в тонкому кишечнику відмічається в результаті зменшення шлункової, панкреатичної секреції і жовчовиділення. Порушення моторної функції /перестальтики/ кишечника і рухливості ворсинок приводять до порушення всмоктувальної функції. Важливу роль в цьому процесі відіграють зміни крово - і лімфообігу, які порушують відтікання продуктів, що всмокталися, і надходження кисню, необхідного для кумуляції енергії.
Всмоктування пригнічується при запальних процесах кишечника, при гіповітамінозах, отруєннях. Внаслідок порушення пристінкового травлення виникають диспепсії, при яких погіршується всмоктування макро- і мікроелементів, що веде до зменшення утворення гемоглобіну і розвитку аліментарної анемії. Зниження всмоктувальної здатності кишечника приводить до нестачі поживних речовин, води, згущених крові.
Порушення моторної функції кишечника проявляється в посиленні, ослабленні, припиненні перестальтики. Посилення рухової функції виникає при запаленні кишечника /ентерит, коліт/, під впливом механічних чи хімічних подразників, погано перетравленої їжі, в результаті дії бактеріальних токсинів, при подразненні блукаючого нерва, після прийому проносних речовин. При посиленні перестальтики прискорюється проходження кормових мас в кишках, погіршується їх перетравлення, всмоктування і розвивається пронос, який сприяє звільненню організму від токсичних чи неперетравних речовин. В той час прискорене виведення з організму води, мінеральних і поживних речовин приводить до виснаження організму.
Ослаблення перестальтики спостерігається при недостатній годівлі, пригніченні блукаючого нерва. Ослаблення перестальтики чи її припинення приводить до застою вмістимого кишечника /в тонкому - кишечнику - хімостаз, в товстому - копростаз,
Синдромом мальдігестії позначають порушення порожнинного травлення, обумовлені недостатнім надходженням у кишечник травних ферментів.
Він виникає внаслідок:
1) недостатності секреторної функції шлунка;
2) недостатності секреції соку підшлункової залози;
3) недостатньої секреції жовчі;
4) недостатньої секреції кишкового соку (12-палої та порожньої кишок).
І. При недостатній секреції соляної кислоти і пепсину у шлунку порушується розщеплення білків, особливо, перетравлення сполучної тканини. Оскільки пілорус постійно відкритий, груба їжа поступає у тонку кишку, подразнює її.
ІІ. Зменшення виділення панкреатичного соку веде до порушення порожнинного травлення. Панкреатичний сік (1,5-2,0 л на добу, має рН – 8,0-8,5 із-за значного вмісту іонів Na+ та бікарбонатів) нейтралізує кислу реакцію хімусу, який надходить із шлунка.
П р и ч и н а м и такого явища можуть бути:
а) нейрогенне гальмування зовнішньосекреторної функції підшлункової залози при зменшенні тонусу блукаючого нерва чи отруєнні атропіном і ін.;
б) дуоденіт, унаслідок чого зменшується утворення стимуляторів панкреатичної секреції : секретину і холецистокінін-панкреозиміну;
в) порушення виведення підшлункового соку (закупорка проток, їх стиснення);
г) зменшення кількості секреторних клітин (руйнування, хронічні панкреатити);
д) спадково обумовлена недостатність ентерокінази, внаслідок чого порушується початкова активація протеолітичних ферментів підшлункового соку: трипсину, хімотрипсину.
Такі фактори ведуть до порушення надходження у 12-палу кишку ферментів підшлункової залози, які відіграють вирішальну роль в травленні білків, жирів і вуглеводів, зокрема:
1) ферментів, які розщеплюють білки (протеаз): трипсину, хімотрипсину, карбоксипептидази А і В, еластази і колагенази;
2) ферментів, які розщеплюють жири: ліпази, фосфоліпаз А1, А2, С, D, лецитинази;
3) ферментів, які розщеплюють вуглеводи: α-амілази, лактази, сахарози;
4) ферментів, які розщеплюють нуклеїнові кислоти (нуклеаз): ДНКази, РНКази.
91. Порушення порожнинного та пристінкового травлення в кишках. Синдром мальабсорбції: визначення поняття, прояви.
Причини недостатності порожнинного травлення
• Захворювання підшлункової залози, як спадкові, так і набуті (хро- нічний панкреатит, стан після панкреатектомії, рак підшлункової залози, муковісцидоз).
• Секреторна недостатність шлунка (атрофічний гастрит, постгастр- ектомічний синдром).
• Дефіцит жовчних кислот або асинхронізм надходження жовчі в тонку кишку при біліарній обструкції, гепатитах, цирозах, ШКХ, після холецистектомії.
• Інактивація травних ферментів при гастродуоденіті, виразковій хворобі ДПК, дисбактеріозі тонкої кишки.
• Порушення транзиту кишкового вмісту та змішування ферментів із хімусом при дуодено- і гастростазі, синдромі подразненого кишечника.
Причини порушення пристінкового травлення
Порушення пристінкового травлення пов'язані з порушенням функції ферментів пристінкового травлення (наприклад, лактазна недостатність).
Порушення пристінкового травлення розвиваються внаслідок диса- харидазної недостатності (вроджена і набута лактазна недостатність); дис- трофічних змін або загибелі ентероцитів (глютенова ентеропатія, саркоїдоз, хвороба Крона, надлишковий бактеріальний ріст).
При недостатності травлення в порожнині кишечника залишається велика кількість недоперетравлених нутрієнтів, що призводить до пору- шення складу внутрішнього середовища кишечника, у тому числі зміни рН, осмотичного тиску, хімічного складу. Зазначені зрушення призво- дять, з одного боку, до вторинного пошкодження слизової оболонки ки- шечника і ще більшого порушення процесів травлення, з іншого – до змі- ни складу мікрофлори кишечника, який посилює наявні порушення.
Синдром мальабсорбції – симптомокомплекс, зумовлений порушенням перетравлення (мальдигестія) і власне всмоктування (мальабсорбція) в тонкій кишці однієї або кількох харчових речовин (здебільшого вуглеводів, білків, а також жирів, мінеральних речовин, вітамінів), що проявляється хронічним проносом; це призводить до тяжких розладів харчування та метаболічних змін. Комплекс змін травлення і всмоктування за міжнародною термінологією об'єднують терміном "мальасиміляція".
Основні симптоми мальабсорбції: пронос, синдром токсикозу, який у подальшому супроводжується ексикозом, гемодинамічними розладами, дистрофією і супутніми дефіцитними захворюваннями (полігіповітаміноз, анемія, рахіт, імунодефіцит).
Синдром мальабсорбції вуглеводів характеризується бродильною диспепсією: рН малооб'ємних пінистих випорожнень нижче ніж 6,0; запах кислий; під час мікроскопічного дослідження виявляють велику кількість крохмальних зерен, клітковини, бродильної флори (дріжджі, клостридії); під час біохімічного дослідження – багато вуглеводів, молочної кислоти.
Синдром мальабсорбції білків характеризується диспепсією гниття: випорожнення мають неприємний запах, лужну реакцію (рН > 7,0), велику кількість неперетравлених м'язових волокон, сполучної тканини, підвищений вміст азоту.
Синдром мальабсорбції жирів характеризується стеатореєю: об'ємні випорожнення жирні, блискучі, тістоподібні, біло-сірі, погано змиваються з пелюшок, містять багато нейтрального жиру, жирних кислот та їх солей.
Найчастішим видом синдрому мальабсорбції є дисахаридазна недостатність. Вона частіше буває вторинною, має транзиторний характер. За етіологією розрізняють лактазну, сахаразну, ізомальтазну недостатність або їх поєднання; вони виникають через недостатність відповідно β-галактозидази, сахарази, ізомальтази
92. Етіологія та патогенез гострого та хронічного панкреатиту. Патогенез панкреатичного шоку .
Гострий панкреатит
Етіологія: 1)жирна їжа, алкоголь 2)жовчні камені та поліпи проток підшлункової 3)травми операції 4)вірус епідпаротиту, коксакі, батер.інф-ї 5)інтоксикації в т.ч.ліками.
Патогенез: активація підшлункового соку в протоках, ос.трипсину, лавоноподібніаутокаталітичні р-ї від активованих ферментів до неактивованих. Місцеві зміни: маоперетравлювання трипсином, хімотрипсином, еластазою, ліпазою, фосфоліпазою з фосфоліпідів арахідонова к-та –простогландини –з крові калікреїноген та альфа2-глобуліни +трипсин – утв.кініни; виклик.вторинну альтерацію –набряк, геморагії, гостре запалення генералізація аж до повного некрозу.
Види: 1)Первинно альтеративний (травма, операція, хімічний ф-р, біологічний вірус бактер., ішемія чи алергія –активація лізосомальних ферментів). 2)Гіпертензивний (підв.тиску в протоках, гіперсекреторний –відносна нед-сть при вжианні жирної їжі алкоголю; обтураційний –камені, пухлини. Пошкодження епітелію протоків активація лізосом.ферм.). 3)Рефлюксний (дуодено-панкреатичний при кишковій непрохідності, розслабленні сф.Одді, активатор –ентерокіназа; жовчно-панкреатичний –жовчні к-ти актив. лізосом.ферменти).
Хронічний панкреатит (ХП) – хронічне запалення підшлункової залози, яке виявляється рецидивуючим чи постійним болем у животі, поєднаним з ознаками екзокринної й ендокринної недостатності і відзначений патологічно незворотною деструкцією паренхіми.
Етіологія хронічного панкреатиту
Хронічна алкогольна інтоксикація,довготривала обструкція ,токсична дія хім.сполук,хрон. гіперкальціємія ,спадковість ,ідіопатичнийхр панкреатит.
Патогенез
Алкоголь---збіл. Генерування вільних радикалів .антиоксидантна недостатність—оксидаційний стрес—первинно-альтернативний мех.
Алкоголь—повторні напади гострого панкреатиту –фіброз і псевдокісти в інтерстиції—здавлювання дрібних проток –обструктивний мех.
Алкоголь—гіперсекреція. Зниж.літостатинів----протеїнові пробки,камені---обтурація дрібних проток –мех. Гіперсекреції-гіпосекреції.
Механізми : первинно-альтернативний – необоротне ушкодж клітин паренхіми,вплив патогенних чинників :
мех.обструктивний , мех.гіпер-гіпосекреції.
Панкреатичний шок це прояв гострого панкреатиту з падінням АТ та генералізованимипор.мікроциркуляції.
Патогенез: 1)Больовий мех-м (набряк залози яка давить на сонячне сплетення, ферменти, кініни та простогландинивплив.на нервові закінчення, ГМ збудження гальмування потім пор.дихання та кровообігу –розв.больовий шок) 2)Гуморальний мех. (ферменти поп.в кров, і якщо не інактивуються альфа1інгібітором протеаз, антитромбіном 3, Альфа2МГ тощо, то актив-ся калікреїн-кінінова с-ма, с-ма згортання та фібринолізу, що викликає генер.розшир.судин, плазморагії, падає АТ, розвив. ДВЗ-с-м)
Синдроми: Гостра арт.гіпотензія –колапс, ДВЗ-с-м, гіпоксичний с-м (при пор.гемодинаміки, пригніч.Дих.центру, гемолізі еритроцитів та інтоксикації).
93.Порушення рухової функції шлунка та кишок .Механізм розвитку основних проявів шлункових та кишкових дискінезій.
Це шлункові та кишкові дискінезії, пор.дефекації. Шлункові дискінезії: 1)гіпертонічна (гіпертонія+ гіперкінезія; причини – груба їжа, шл.гіперсекреція, пер.вагуса, мотиліну; веде до затримки вмісту, до підвищ.секреції і виразок, також до розвитку антиперистальтики а отже до відрижки, нудоти, блювоти; відносять також пілороспазм – часто у дітей після внутрішньоутроб.гіпоксії) 2)гіпотонічна (гіпотонія-кінезія; причини – жирна їжа, гіпоацидні гастрити, змен.вагуса, підвищ. ГИП та секретину, видалення воротаря; веде до пор.перетрав. проносів). Кишкові дискінезії: 1)гіперкінетична (причини – збудж.рец. при ентериті коліті, інтоксикації, при ахілії, зб.вагуса, мотиліну. Веде до діареї, пор.травл. та всмоктування, зневоднення, виділь.негаз.ацидоз) 2)гіпокінетична (мало клітковини, шл.гіперсекреція, вікові зміни, змен.вагуса, хв.Гіршпрунга; веде до атонічних закрепів, кишк.аутоінтоксикації, метеоризму та киш.непрохідності). Блювота – складнорефлекторний акт, збудж.блюв.центру у довг.мозку. Стадії: 1)глиб.вдих, опущ.діафрагми, надгортаннику, підняття мякого піднебіння 2)скор.воротаря, тіла та розсл.страв.сф., стравохід розшир.іскор. 3)сильне скор.діафрагми та чер.мязів, виверження. Є три варіанти: 1)центральна (менінгоенцефаліти, пухлини, умовнорефлекторна, вестибулярна) 2)гематогенно-токсична (отрути, СО2, алкоголь, прод.метаболізму вплив на хеморец.блюв.центру) 3)вісцеральна (з вн.органів, шлунку, глотки, вінц.судин, очеревини, жовчних прот.). Пронос 5 типів: 1)осмотична діарея (р-ни які не всмокт.у кишках, послабл., при мальабсорбції та мальдигестії) 2)секреторна (секреція електролітів в просвіт разом з водою, при холері) 3)при гальмув.акт. транспорту іонів (спадк.хлордіарея) 4)при підв.проникностікиш.стінки (запалення) 5)при пор.моторики.
94. Кишкова непрохідність : види ,етіологія ,патогенез.
Кишкова непрохідність – пор.проходження вмісту кишок внаслідок обтурації, передавлення чи пор.ф-й.
Причини та види: 1)механічна а)обтураційна (пухлина, калові камені, гельмінти) б)странгуляційна (заворот, защемлення грижі). 2)динамічна а)спастична б)паралітична.
Патогенез змін: 1)больовий синдром (вн.спазму, розтягнення чи некрозу – аж до больового шоку) 2)зневоднення (обтурація – багато рідини вище – розтягнення ст. –зб.секреції залоз та води – подразнення рец. – блювота). 3)пор.обміну травних ферментів (тиск закидає вміст до проток підшлункової –вин. гострий панкреатит та шок) 4)пор.КОС (блювота, хлориди –негазовий алкалоз, гідрокарбонати –ацидоз) 5)кишк.аутоінтоксикація (проявляється ознаками нед.печінки). 6)гострий перитоніт (м/о прон.черезнекрот.стінку, викликає та підсилює інші ф-ри). 7)пор.гемодинаміки та мікроциркуляції (змен.АТ, ХОК внасл.бол.тапанкр.шоку та зневоднення) 8)гіпоксія (пор.гемодинаіки та дихання) 9)пор.ф-й органів (гіпоксія та інтоксикація).
95. Етіопатогенез некротичного ентероколіту у новонароджених.
Некротичний ентероколіт (НЕК) – захворювання, що характеризується запаленням та розвитком різного ступеня деструктивно-некротичних змін стінки тонкої та/або товстої кишки у дітей перших місяців життя, частіше – в період новонародженості, у недоношених та дітей із наднизькою масою тіла.
Етіологія
НЕК – поліетіологічне захворювання, в основі якого первинно
лежить незрілість шлунково-кишкового тракту. Класично прийнято розглядати
тріаду основних причин, які сприяють розвитку НЕК, серед яких
гіпоксія/асфіксія, перенесена в перинатальному періоді, неадекватне ентеральне
вигодовування в постнатальному періоді, бактеріальна колонізація кишечника
патогенною мікрофлорою.
Захворювання часто спостерігається у недоношених дітей, що пов’язано з великою
частотою серед цієї групи дітей із внутрішньоутробною гіпоксією й асфіксією під
час пологів, особливостями формування мікробіоти кишечника на тлі
антибактеріальної та інтенсивної терапії, незрілістю механізмів нервової
регуляції моторної функції кишечника, порушеннями адаптації кишечника до
ентерального харчування, відсутністю раннього вигодовування грудним молоком,
недосконалістю місцевого імунітету.
96. Недостатність печінки – визначення поняття , клаасифікація недостатності печінки за різними принципами класифікації.Значення здатності органу до регенерації.
Недостатність печінки – це пат.стан, при якому діяльність цього органу не здатна забезпечити постійність гомеостазу орг.відповідно до потреб.
Причини: фізичні ф-ри, хімічні, інфекційні, аліментарні, алергічні, пор.кровообігу, едокринні та обмінні пор., пухлини, генетичні дефекти метаболізму.
Класифікація: 1)відносна (підвищення навантаження, може переходити в абсолютну) та абсолютна (первинне ураження).
2)за етіологією: печінково-клітинна, холестатична та печінково-судинна.
3)за к-стю пор.ф-й: тотальна та парциальна.
4)за клінічним перебігом: гостра та хронічна.
Проявляється недостатність порушенням таких ф-й печінки: 1)метаболічна (БЖВ, віт, гормони) 2)захисна (фагоцитарна, антитоксична) 3)екскреторна (жовч) 4)гемодинамічна.
Клінічні прояви: 1)печінково-клітинна (гіпопротеїнемічний синдром, геморагічний синдром, синдром гепатоцеребральноїнед-сті аж до коми та ін. 2)при пор.екскреторної ф-ї (жовтяниця, холемічний синдром, ахолічний синдром). 3)печінково-судинна нед-сть (синдром портальної гіпертензії).
97. Визначення та поняття цирозу печінки. Причини, компенсаторні механізми, стадії розвитку та типові прояви.
Цироз печінки. Поняттям цироз позначають хронічне прогресуюче ураження печінки, що характеризується, з одного боку, розростанням сполучної тканини, а з другого - патологічною регенерацією печінкової паренхіми (утворенням вузлів регенерації), внаслідок чого змінюється звичайна архітектоніка органа, поділ його тканини на правильні структурні одиниці - печінкові часточки
Розвиток наведених вище змін супроводжується перебудовою судинного русла печінки, порушенням кровопостачання осередків ще життєздатної паренхіми.
Цироз є наслідком необоротного ушкодження й загибелі великої кількості печінкових клітин, його розвитку часто передують стеатоз печінки і гепатити
Залежно від причин ушкодження печінкової паренхіми розрізняють три патогенетичні варіанти цирозу
1) постнекротичний. Розвивається як наслідок некрозу великої кількості гепатоцитів. Клінічним його еквівалентом є гепатоцелюлярна недостатність печінки
2) біліарний. Формується внаслідок тривалого холестазу — порушень виведення жовчі по внутрішньопечінкових і позапечінкових жовчовивідних шляхах (наприклад, при обтурації останніх). Недостатність печінки, яка розвивається при цьому, позначають як холестатичну;
3) портальний. Є наслідком хронічних порушень кровообігу в печінці (наприклад, при застійній недостатності серця). Клінічно виявляє себе ознаками печінкової недостатності, що її відносять до гепатоваскулярного варіанта.
98. Етіологія та патогенез печінкової недостатності. Типові прояви (синдроми).
Етіологія: Печінкова недостатність виникає при :
· вірусних гепатитах (А і В),
· гострих отруєннях (грибами, дихлоретаном, фосфором, чотирьоххлористим вуглецем, миш’яком),
· важких токсикозів вагітності,
· опікової хвороби,
· застосування інгаляційних анестетиків, антибіотиків чи сульфаніламідних середників з гепатотоксичною дією,
· при масивній бактеріальній інвазії,
· цирозах,
· первинних та метастатичних пухлинах печінки.
Патогенез: Розрізняють три основні механізми ушкодження гепатоцитів і відповідно три патогенетичні варіанти абсолютної печінкової недостатності:
1. Пряме ушкодження печінкових клітин патогенними агентами - печінково-клітинна (гепатоцелюлярна) недостатність.
2. Ушкодження гепатоцитів, зумовлене тривалим холестазом, — холестатична недостатність печінки.
3. Первинні порушення кровообігу в печінці - печінково-судинна (гепатоваскулярна) недостатність. Основним механізмом ушкодження гепатоцитів у цих випадках є гіпоксія.
Типові прояви:
У вигляді:
1. Екскреторної форми (порушення виділення жовчі, характерна ознака - жовтяниця).
2. Васкулярної форми (переважає клініка портальної гіпертензії).
3. Печінково-клітинної форми (порушення діяльності гепатоцитів із розладами різноманітних функцій печінки).
По перебігу розрізняють гостру і хронічну печінкову недостатність, по ступеню компенсації - компенсовану, субкомпенсовану та декомпенсовану.
Ураження ЦНС перебігає стадійно: прекома, загрожуюча кома та власне кома.
Клінічні прояви: жовтяниця, судинні зірочки, “пальмарна” долоня, розширення дрібних поверхневих судин обличчя,лихоманка, печінковий запах з рота, від поту та сечі, розлади травлення,дихальна недостатність, розлади діяльності серцево-судинної системи,геморагічний синдром, анемії, ниркова недостатність; гепато-ренальний синдром, симптоми інтоксикації центральної нервової системи, прогресивне швидке зменшення розмірів печінки.
99. Експериментальні моделі недостатності печінки. Фістули Екка, Екка-Павлова, ангіостомія за Лондоном.
Печінково-клітинна недостатність:
· повне видалення печінки;
· часткове видалення печінки;
· токсичне ураження печінки гепатотропними отрутами (чотирихлористий вуглець, хлороформ, фосфор та ін.)
Холестатична недостатність:
· перев’язування жовчовивідних протоків
· Печінково-судинна недостатність:
· накладання порто-ковальних анастомозів (фістула Екка, фістула Екка-Павлова);
· перев’язування ворітної вени;
· перев’язування печінкових вен;
· накладання лігатури на печінкову артерію.
Класичними методами дослідження печінкових функцій і відтворення печінкової недостатності стали хірургічні втручання, метою яких є моделювання порушень кровообігу в печінці та повне видалення цього органа. До таких методів можна віднести:
Фістула Екка - полягає в наступному: фістула накладається між ворітньою і задньою порожнистою веною, з послідуючим перев’язуванням вени вище сполучення. При цьому кров з шлунково-кишкового тракту в печінку не поступає, а йде в велике коло кровообігу. У тварини спостерігається отруєння аміаком, який в печінці перетворюється в сечовину Цей експеримент дозволив вивчити знешкоджуючу , а також сечоутворюючу функції печінки.
Фістула Екка-Павлова- накладують порто-кавальний анастомоз, але перев’язують нижню порожнисту вену. При цюому вся кров йде в печінку, розвиваються периферичні колатералі – голова медузи. Перев’язують воротну вену вище анастомозу. Вважали, що тварини гинули від гіпоглікемії, якщо їх тримати на безбілковій дієті. Але навіть при введенні в/в глюкози смерть наставала через 24 години. Вважають, що навіть 10% повноцінної печінки можуть забезпечувати виконання її функції.
А нгіостомія по Е.С. Лондону - спосіб накладання на кровоносну судину фістули, за допомогою якої періодично у тварини беруть кров для дослідження в умовах хронічного досліду. Для цього до стінок ворітньої і печінкової вен підшивають фістулу, з виходом назовні, що дозволяє брати кров, яка надходить і відтікає від печінки.
100. Порушення вуглеводного, білкового та ліпідного обміну при недостатності печінки. Клінічні прояви та їх механізми.
Порушення вуглеводного обміну при ураженні печінки полягає у зміні таких процесів: 1) розщеплення і синтезу глікогену; 2) окиснення глюкози;. 3) гліконеогенезу; 4) перетворення галактози і фруктози в глюкозу; 5) утворення глюкуронової кислоти. Ці порушення можуть мати набутий і спадковий характер.
Основним механізмом виникнення цих порушень є зниження активності ферментів, що каталізують різні ланки вуглеводного обміну, внаслідок зменшення синтезу їх при білковому голодуванні, дефіциті енергії у зв'язку з гіпоксією, ушкодженні мітохондрій гепатоцитів, спадкових ферметопатіях, розладі нейрогуморальної регуляції вуглеводного обміну.
Порушення вуглеводного обміну проявляється розвитком гепатогенної гіпоглікемії, спадковими захворюваннями — глікогенозами, галактоземією, фруктозуріею. Гіпоглікемія зумовлена зменшенням вмісту глікогену в печінці, зниженням глікогенолізу (наприклад, при глікогенозах Гірке і Герса) та гліконеогенезу (при аддісоновій хворобі, коли падає секреція глікокортикоїдів). Зниження вмісту глікогену в печінці спричинює ослаблення її знешкоджувальної функції внаслідок порушення перетворення глікогену в глюкуронову кислоту.
Характерними ознаками порушення ліпідного обміну при захворюваннях печінки є: 1) дефекти розщеплення і всмоктування жирів їжі в кишках (у разі патології жовчотворення і жовчовиділення у зв'язку з дефіцитом жовчних кислот); 2) порушення синтезу й окиснення тригліцеридів, фосфоліпідів, ліпопротеїдів, холестерину; 3) збільшене утворення кетонових тіл.
Розлад жирового обміну в печінці спричинює розвиток жирового гепетозу (жирова дистрофія, жирова інфільтрація печінки), під час якого в гепатоцитах накопичується жир і відбувається дифузне або осередкове ожиріння печінки. Причинами виникнення жирового гепатозу є аліментарні фактори (голодування, особливо білкове, нестача в їжі ліпотропних речовин — холіну, метіоніну, надлишок вуглеводів і жирів); токсичні речовини (алкоголь, гепатотропні отрути — інсектициди, тетрациклін у великих дозах); ендокринні й метаболічні порушення (цукровий діабет, ожиріння); гіпоксія (при недостатності серця, дихання).
Патологічні процеси в печінці (гепатит, цироз) нерідко супроводжуються зменшенням утворення естерифікованого холестерину або зниженням загальної кількості його в крові, порушенням синтезу й окиснення холестерину, перетворення його в жовчні кислоти і виведення з жовчю.
Порушення білкового обміну при патології печінки проявляється розладом таких процесів: 1) синтезу білків (у тому числі білків плазми крові); 2) розщеплення білків (до амінокислот, пуринових і піримідинових основ); 3) дезамінування, трансамінування та декарбоксилування амінокислот; 4) утворення сечовини, сечової кислоти, аміаку, глутаміну (транспортної форми аміаку в крові), креатину — кінцевих продуктів білкового обміну.
Розрізняють такі механізми порушення білкового обміну в печінці:
ушкодження внаслідок патологічного процесу (гепатит, гепатоз, цироз, пухлина, ішемія) печінкових клітин як структурного субстрату анаболізму і катаболізму білків;
порушення генетичної регуляції синтезу білків внаслідок ушкодження структурних генів, рибосом цитоплазми і гранулярної
ендоплазматичної сітки гепатоцитів, дефіциту РНК, що спричинює зміну кількості синтезованих білків, утворення аномальних за своєю структурою білків (наприклад, при амілоїдозі печінки, спадковій афібриногенемії);
дефіцит амінокислот (при білковому голодуванні, порушенні перетравлювання і всмоктування білків у кишках);
дефіцит енергії (в разі гіповітамінозу, особливо дефіциту піридоксину, рибофлавіну; при гіпоксії);
порушення нейрогуморальної регуляції білкового обміну (наприклад, при дефіциті інсуліну, зміні секреції соматотропіну аденогіпофізом).
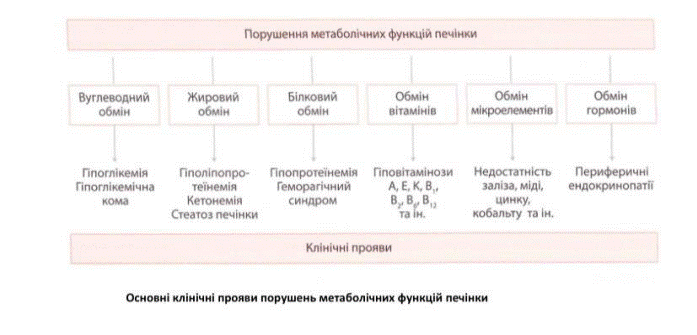
101.Порушення дезінтоксикаційної функції печінки. Види, етіологія та патогенез печінкової коми. Роль церебротоксичних речовин у розвитку печінкової енцефалопатії.
Антитоксична функція властива гепатоцитам. Вона полягає в інактивації кінцевих продуктів обміну речовин (сечовиноутворення) і екзогенних сполук, що надходять з кишок (продукти діяльності мікрофлори - фенол, крезол, індол, скатол, аміни), а також з навколишнього середовища. Детоксикація різних речовин здійснюється за допомогою різних хімічних реакцій (синтезу, окиснення, відновлення, гідролізу, метилювання, кон’югації), що відбуваються в структурах ендоплазматичного ретикулуму (мікросомах), мітохондріях, пероксисомах і цитоплазмі печінкових клітин. До порушень антитоксичної функції печінки можуть спричинятися:
1) зменшення кількості функціонуючих гепатоцитів (наприклад, при масивних некрозах печінки). Так, установлено, що синтез сечовини з аміаку порушується при ураженні 80 % і більше паренхіми печінки;
2) зменшення активності ферментів, що беруть участь у реакціях детоксикації. Воно може бути зумовлене генетичними дефектами відповідних ферментів (наприклад, спадкове порушення ферментів синтезу сечовини) або набутими порушеннями їх утворення (розлади білоксинтезуючої функції печінки);
3) зменшення концентрації речовин, що беруть участь в інактивації токсичних продуктів (кисню, глюкуронової та сірчаної кислот, гліцину, таурину, цистеїну тощо);
4) дефіцит АТФ. При цьому можуть порушуватися процеси надходження й виведення з гепатоцитів токсичних речовин, а також реакції синтезу й кон’югації, які вимагають витрат енергії.
Печінкова кома. Печінковою комою називають патологічний стан, що виникає в результаті тяжких порушень антитоксичної функції печінки і виявляє себе втратою свідомості, випадінням рефлексів на внутрішні й зовнішні подразники, розладами життєво важливих функцій організму (кровообігу, дихання). Печінкова кома є крайнім
проявом синдрому гепатоцеребрсиїьної недостатності, а тому має ті ж самі причини, що і цей синдром. Залежно від джерела й механізмів надходження в кров церебротоксичних речовин виділяють два патогенетичних варіанти печінкової коми:
I. Ендогенну {печінково-клітинну, або розпадну). У цьому випадку поява церебротоксичних речовин у крові пов’язана з порушеннями антитоксичної функції печінки при ушкодженні й загибелі гепатоцитів;
II. Екзогенну {портокавальну, або шунтову). Її розвиток обумовлений тим, що церебротоксичні речовини потрапляють у системний кровообіг з кишок через ворітну вену та портокавальні анастомози, минаючи печінку. При цьому антитоксична функція печінки може істотно не страждати. Центральне місце в патогенезі печінкової коми належить надходженню в кров аміаку та інших церебротоксичних речовин
Механізми порушень діяльності центральної нервової системи, що ведуть до печінкової коми, досить складні. Проте можна виділити ряд провідних патогенетичних чинників. L Порушення діяльності синапсів і синоптичної передачі. Вони виявляють себе послабленням процесів центрального збудження і посиленням центрального гальмування.
Серед причин наведених змін такі: а) дія аміаку. б) несправжні нейромедіатори (псевдонейромедіатори). в) дія гальмівних медіаторів
.Роль церебротоксичних речовин у розвитку печінкової енцефалопатії.
Порушення антитоксичної функції печінки виявляють себе ознаками інтоксикації, що уражує насамперед центральну нервову систему. Тому комплекс змін, що виникають, одержав назву синдрому гепатоцеребральної недостатності, відомого ще як печінкова енцефалопатія. Для нього характерними є різні поєднання психічних і невротичних розладів аж до втрати свідомості й розвитку коматозного стану. Залежно від ступеня порушень антитоксичної функції печінки зазначений синдром може виявляти себе:
а) емоційно-психічними розладами: емоційною нестійкістю (почергові зміни ейфорії й депресії), безсонням уночі й сонливістю вдень, головним болем, запамороченнями;
б) вираженими порушеннями свідомості - розвитком ступору (сонливість, сплутаність свідомості);
в) печінковою комою (див. нижче).
В основі синдрому гепатоцеребральної недостатності лежить накопичення в крові так званих церебротоксичних речовин. Серед них особливо важливого значення надають таким сполукам:
1) аміаку. В організмі існує два джерела надходження аміаку в кров:
(а) усі периферичні, де відбуваються процеси дезамінування амінокислот, і
(б) кишки, де утворення аміаку обумовлене процесами гниття, тобто діяльністю кишкової мікрофлори. Аміак є токсичною сполукою, а тому в нормі його в крові практично немає, бо він перетворюється в гепатоцитах у сечовину. Із кишок аміак надходить одразу в печінку по судинах портальної системи, а з периферичних клітин — у зв’язаній формі у складі глютаміну (т. зв. глютаміновий човниковий механізм транспорту NH3 ). При недостатності печінки аміак, що надходить сюди із двох зазначених джерел, не може повністю перетворюватися в сечовину, а тому його концентрація в крові зростає;
2) продуктам окиснення амінокислот у кишках. Такими є фенол, індол, скатол, аміни (путресцин, кадаверин); сполуки сірки, що утворюються при окисненні метіоніну (диметил сульфід, метил меркаптан). З останніми, зокрема, пов’язують появу специфічного симптому печінкової коми - печінкового запаху,
3) низькомолекулярним жировим кислотам: масляній, валеріановій, капроновій. Вони з’являються внаслідок порушень окиснення жирових кислот в ушкоджених гепатоцитах.
102. Порушення жовчоутворювальної та жовчовидільної функції печінки. Види жовтяниць. Їх етіологія та патогенез.
Порушення жовчоутворювальної функції печінки виявляється у збільшенні чи зменшенні секреції жовчі, як правило, з одночасною зміною складу її.
Етіологія. Порушення жовчоутворення можуть спричинити: 1) зміна нейрогуморальної регуляції; 2) аліментарні фактори (жири, яєчний жовток, білкове голодування) деякі лікарськи рослини і препарати (настій кукурудзяних приймочок, сорбіт тощо); 3) екзогенні та ендогенні фактори, що порушують енергетичний обмін в організмі (гіпоксія, перегрівання, гіпотермія, отруєння ціанідами) 4) ураження печінки і жовчних шляхів.
Патогенез. До механізмів, що зумовлюють якісні й кількісні порушення жовчоутворювання, належать: 1) зміна секреторної активності гепатоцитів; 2) порушення реабсорбції компонентів жовчі у жовчних шляхах і кишках (печінково-кишковий кругообіг); 3) зміни транс- та інтерцелюлярної фільтрації деяких речовин з крові в капіляри печінки
Порушення жовчовиділення
Етіологія. Причинами порушення надходження жовчі в дванадцятипалу кишку можуть бути: 1) механічна перешкода відтоку жовчі - здавлення жовчних шляхів іззовні (пухлиною головки підшлункової залози, запаленою тканиною, рубцем) або закупорка її (каменем, гельмінтами, густо. Жовчю); 2) порушення іннервації жовчних шляхів – гіпер- або гіпокінетична дискінезія (наприклад, зменшення жовчовиділення при спазмі сфінктера шийки жовчного міхура); 3) зміна гуморальної регуляції жовчовиділення.
Патофізіологічні синдроми, зумовлені порушенням жовчоутворення і жовчовиділення супроводжуються жовтяницею.
Види жовтяниці.
1. Гемолітична (надпечінкова) жовтяниця.
Етіологія. Виникає в результаті гемолізу еритроцитів і збільшеного утворення білірубіну в клітинах системи мононуклеарних фагоцитів.
Патогенез. Внаслідок посиленого гемолізу еритроцитів у зірчастих ретикулоедотеліоцитах, макрофагах селезінки, кісткового мозку утворюється така велика кількість непрямого білірубіну, що гепатоцити печінки не здатні повністю вилучити його з крові і зв’язати з уридиндифосфоглюкуроновою кислотою (відносна недостатність печінки). У крові збільшується вміст непрямого білірубіну (непряма гіпербілірубінемія), який не виводиться з сечею, оскільки зв’язаний з альбуміном плазми. При гемолітичній жовтяниці в печінці, жовчних шляхах та кишках синтезується надлишкова кількість глюкуронідів білірубіну.
2. Паренхіматозна (печінкова) жовтяниця.
Етіологія. Її розвиток пов'язаний з ураженням печінки.
Патогенез. Зменшене виділення білірубіну поєднується з порушенням його захвату, внутрішньоклітинного транспорту і кон’югації.
Такий механізм виникнення жовтяниці у разі ушкодження клітин печінки (гепатоцелюлярна жовтяниця) і внутрішньопечінкового холестазу (холестатична жовтяниця).
При гепатоцелюлярній і холестатичній печінковій жовтяниці - різко знижується секреція прямого білірубіну в жовч, він надходить з патологічно змінених гепатоцитів у кров, виникає пряма гіпербілірубінемія. Водночас у крові підвищується рівень непрямого білірубіну - непряма гіпербілірубінемія, що пов’язано зі зниженням його захвату, внутрішньоклітинного транспорту й утворення глюкуронідів білірубіну. Потрапляння у кров жовчних кислот. Зменшення або припинення надходження жовчі у кишки (гіпохолія, ахолія) призводить до зниження утворення метаболітів білірубіну та виділення їх з калом і сечею (сліди стеркобіліну), а також появи ахолічного синдрому.
3. Механічна (обтураційна, або підпечінкова) жовтяниця.
Етіологія. Виникає в результаті порушення відтоку жовчі по жовчовивідних шляхах.
Патогенез. Механізм перешкода відтоку жовчі спричинюється до застою (поза печінковий холестаз) і підвищення тиску жовчі понад 2,7 кПа (270 мм вод.ст..), розширення і розриву жовчних капілярів та надходження жовчі прямо у кров або через лімфатичні шляхи. Поява жовчі в крові зумовлює пряму гіпербілірубінемію, гіперхолестеринемію, розвиток полемічного синдрому, білірубінемію (звідси темно-жовте забарвлення сечі - "колір пива") і наявність жовчних кислот і сечі.
103. Порушення обміну жовчних пігментів при різних видах жовтяниць: вміст білірубіну та його фракцій, уробіліногену і стеркобеліну у крові та сечі.
Гемолітична (надпечінкова) жовтяниця.
а) збільшення вмісту непрямого білірубіну в крові; б) збільшення вмісту стеркобіліногену в калі (гіперхолічний кал); в) збільшення вмісту стеркобіліногену в сечі; г) поява в сечі уробіліногену (у зв'язку з тим, що печінка не в змозі окислити великі кількості цієї речовини, яка надходить із кишок).
Паренхіматозна (печінкова) жовтяниця.
а) збільшення вмісту непрямого білірубіну в крові; б) збільшення вмісту стеркобіліногену в калі (гіперхолічний кал); в) збільшення вмісту стеркобіліногену в сечі; г) поява в сечі уробіліногену (у зв'язку з тим, що печінка не в змозі окислити великі кількості цієї речовини, яка надходить із кишок).
Механічна (обтураційна, або підпечінкова) жовтяниця.
а) збільшується вміст у крові прямого білірубіну (гіпербілірубінемія); б) у крові з'являються жовчні кислоти (холалемія); в) збільшується вміст у крові холестеролу (гіперхолестеролемія). З'являються моди- j фіковані ліпопротеїди (ліпопротеїд X), що мають атерогенні властивості; г) у сечі з'являється білірубін (білірубінурія), унаслідок чого вона набуває темного забарвлення ("колір пива"), крім того в ній виявляють жовчні кислоти (холалурія). Із сечі зникає стеркобіліноген; ґ) у калі нема стеркобіліногену (безбарвний кал). Зазначені зміни пігментного обміну зумовлюють розвиток двох важливих клінічних синдромів, характерних для механічної жовтяниці: холемічного і ахолічного.
104. Дія жовчних кислот на організм. Холемічний та ахолічний синдроми при різних видах жовтяниць. Етіопатогенез фізіологічної жовтяниці новонароджених
Холємічний синдром (синдром холестазу) обумовлений надходженням компонентів жовчі (жовчних кислот, прямого білірубіну, холестеролу) у кров у зв'язку з порушенням формування і відтоку жовчі. Він закономірно виникає при механічній жовтяниці, а також деяких формах печінкової жовтяниці (печінково-клітинній, печінковій жовтяниці, обумовленій порушеннями екскреції жовчі).
Ахолічним називають синдром, обумовлений ненадходженням жовчі в кишки у зв'язку з порушеннями її формування і відтоку. Внаслідок відсутності в кишках жовчних кислот не активується ліпаза, не емульгуються жири, не утворюються розчинні комплекси жовчних кислот із жирними кислотами, у зв’язку з чим 60-70 % жирів не перетравлюються, не всмоктуються і видаляються з організму разом із калом (стеаторея). При ахолії знижується ферментативна дія трипсину й амілази, погіршується перетравлювання білків і вуглеводів, знижується тонус і перистальтика кишок – з’являються закрепи. Кал знебарвлений – відсутність стеркобіліну, який не утворюється в кишках через припинення екскреції глюкуронідів білірубіну, отже, не міститься й у сечі.
Етіопатогенез фізіологічної жовтяниці новонароджених. Жовтяниця виникає через посилене відкладання в шкірі білірубіну, продукту розпаду гемоглобіну(механізми зв’язування і виведення білірубіну дозрівають через дві неділі після народження). Майже у кожного новонародженого розвивається транзиторна помірна некон’югаційна гіпербілірубінемія(фізіологічна жовтяниця). Відкладання, як правило, досягає максимуму на п'яту добу життя, після чого поступово зменшується.
105. Патогенез білірубінової енцефалопатії новонароджених («ядерної жовтяниці»): механізми нейротоксичності білірубіну.
Ядерна жовтяниця та механізм.
Не зв’язанний с альбуміном білірубін може проникати до тканин, особливо в тканину головного мозку грудних дітей, надаючи токсичну дію. Концентрація незв’язанного білірубіну може зростати при тяжких захворюваннях системи крові чи в тих випадках, коли лікувальні засоби, що зв’язують альбумін, витісняють білірубін у зв’язку з білком. Гемолітична хвороба новонароджених веде до накопичення в тканинах головного мозку некон’югованого білірубіну, здатного серйозно нашкодити головному мозку. Таке пошкодження називають ядерною жовтухою. Всупереч, кон’югований білірубін розчинний у воді, нетоксичний і в дуже малій степені зв’язується з альбуміном. Через гарну розчинність і погане зв’язування с альбуміном надлишок кон’югованого білірубіну може бути виведений із мочею. При довготривало підвищеній концентрації кон’югованого білірубіну частина його(дельта-фракція білірубіну) може ковалентно зв’язуватись з альбуміном
При наявності факторів ризику і значному підвищенні рівня білірубіну виникає ризик потрапляння його з крові в тканини головного мозку і стійкого пошкодження там особливо чутливих структур — базальних ядер (базальних гангліїв). Розвивається ядерна жовтяниця.
106. Дисхолія. Жовчокам’яна хвороба. Фактори утворення жовчних каменів.
Дисхолія - це порушення фізико-хімічних властивостей жовчі, внаслідок чого вона набуває літогенних властивостей, тобто здатності утворювати камені (конкременти) у жовчному міхурі й жовчних протоках. Результатом цього є розвиток жовчноком 'яної хвороби.
Жовчнокам'яна хвороба - захворювання, яке виникає внаслідок утворення каменів(конкрементів) у жовчному міхурі(холецистолітіаз), рідше — у внутрішньопечінкових жовчних протоках, ще рідше — у загальній жовчній печінковій протоці.
Фактори: а)спадкова схильність;
б)нераціональне харчування;
в)порушення обміну речовин;
г)інфекційно-запальні процеси в жовчному міхурі і жовчних протоках;
ґ) застій жовчі (холестаз)
107. Синдром портальної гіпертензії: етіологія, експериментальне відтворення, патогенез, прояви. Механізми розвитку асциту.
Портальна гіпертензія - це підвищення тиску крові в басейні ворітної вени, що клінічно проявляється такими симптомами як розширення портокавальниханастомозів, асцитом, спленомегалією та кровотечами з варикознорозширенихколатералей.
Етіологія
1.Надпечінкова ПГ:
• збільшення портального венозного кровотоку, що
спостерігається за наявності артеріовенозної фістули, спленомегалії, не
пов'язаної із захворюваннями печінки, каверноматозом ворітної вени;
• тромбоз або оклюзія портальних або селезінкових
вен.
2. Внутрішньопечінкова ПГ – захворювання
печінки:
• гострі: алкогольний гепатит, фульмінантний
вірусний гепатит;
• хронічні: алкогольний та неалкогольний
стеатогепатит, хронічний гепатит помірної та високої активності, вірусний,
токсичний, медикаментозний,
аутоімунний, первинний біліарний ЦП, ЦП
будь-якої етіології, ураження печінки при хворобах накопичення
(Вільсона-Коновалова, гемохроматоз, недостатність α1-антитрипсину,
вроджений фіброз печінки, шистозоматоз, саркоїдоз, метастатична гіперплазія
печінки, фокальна вузликова гіперплазія печінки.
3. Постпечінкова ПГ:
• захворювання печінкових венул і вен, нижньої
порожнистої вени: вроджене мембранозне зрощення нижньої порожнистої вени,
венооклюзивна хвороба, тромбоз печінкових вен (хвороба та синдром Бадда-Кіарі),
тромбоз, дефекти розвитку нижньої порожнистої вени, пухлинне стискання нижньої
порожнистої або печінкової вен;
• захворювання серця: кардіоміопатія,
захворювання серця з ураженням клапанів, констриктивний перикардит.
Патогенез
Основним патогенетичним механізмом є механічна перешкода відтоку крові, зумовлена тромбозом, облітерацією або стисканням із зовні великих судин (ворітної вени, печінкових вен).
Прояви
· Збільшенийрівень тиску у ворітній вені.
· Рівень внутрішньоселезінкового тиску вище 17 мм рт. Ст.
· Метеоризм, відчуття переповнення кишечнику, біль у животі без чіткої локалізації, зниження апетиту, чергування запорів з проносами, збільшення живота, ознаки дефіциту харчування.
Механізми розвитку асциту
підвищений тиск y системі ворітної вени, гіпоальбумінемія, надмірна продукція рідини, порушення відтоку лімфи.
108. Причини та механізми порушення клубочкової фільтрації, канальцевої реабсорбції та секреції. Використання функціональних проб для визначення стану основних функцій нирок.
Порушення клубочкової фільтрації
· зниження ефективного фільтраційного тиску при гіпотензивних станах (гіпотензії, колапсі та ін.), ішемії нирки (нирок), гіповолемічних станах;
· зменшення площі клубочкового фільтрату; спостерігається при некрозі нирки (нирок) або її частини, мієломній хворобі, хронічних гломерулонефритах та інших станах;
· зниження проникності фільтраційного бар'єру внаслідок потовщення, реорганізації базальної мембрани або інших її змін; відбувається при хронічних гломерулонефритах, цукровому діабеті, амілоїдозі та інших хворобах
· збільшення проникності фільтраційного бар'єра (наприклад, внаслідок розпушення базальної мембрани) під впливом БАВ-медіаторів запалення або алергії (гістаміну, кінінів, гідролітичних ферментів).
Порушення канальцевої реабсорбції
при різних ферментопатіях і дефектах систем трансепітеліального перенесення 6 речовин (наприклад, амінокислот, альбуміну, глюкози, лактату, бікарбонатів та ін.), а також мембранопатій епітелію і базальних мембран ниркових канальців.
Порушення секреції
при генних дефектах і призводять до цистинурії, аміноацидурії, фосфатурії, печінкового діабету, бікарбонатурії, ниркового ацидозу.
Проба Зимницького. Дає можливість констатувати ранні порушення концентраційної функції нирок.
Метод кліренсу - дозволяє оцінити здатність нирок очищати кров.
Метод Реберга дозволяє оцінити функцію клубочкової фільтрації і канальцевої реабсорбції нирок.
109. Кількісні та якісні зміни складу сечі. Олігурія, анурія та поліурія. Водний, осмотичний та гіпертензивний діурез. Гіпо- та ізостенурія.
Якісні зміни сечі
Сеча здорової людини прозора, світло-жовтого кольору. Реакціясечі в норміслабокисла, її pH коливаєтьсявід 5,0 до 7,0 і залежитьвід характеру харчування.
Кількісні зміни сечі
В нормі добовий діурез повинен складати близько 1,5 л, денний діурез – в 3 рази перевищувати нічний, значення питомої ваги хоча б в одній із порцій не має бути меншим 1022, різниця між мінімальним та максимальним значенням питомої ваги має бути значною (не менше 10).
Олігурія – це зменшення діурезу нижче облігатногообєму, тобто 700мл/добу. Анурія –це повна відсутність діурезу. Причиною олігуріії є пор.клуб. фільтрації коли ШКФ стає нижче 10мл/хв. Веде до гіпергідрії, гіпернатрійкалійємії, азотемії.
Поліурія – це зб.діурезу вище 1,8л. Причини:позаниркові (психогенна полідипсія, пор.водно-сольового обміну, напр.нецукровий діабет) та ниркові (поліурична стадія Г та ХНН).
Види поліурії: 1)водний діурез (змен.факультативної реабсорбції води при водному навантаженні, нецукровому діабеті. Сеча гіпотонічна).
2)осмотичний діурез (салурез, при пор.реабсорбції електролітів, підвищенні вмісту порогових р-н у первинній сечі напр.приц.діабеті, дії поганореабсорбуючих р-н та салуретиків.
3)гіпертензивний діурез (АГ, прискорення кровотоку у прямих судинах мозкового шару і збільшення конвекційного транспорту – вимивання На, СІ сечовини з інтерстицію – зменшується осмотичний тиск позаклітинної рідини і змен.реабс.води).
Гіпо-ізостенурія. Гіпостенуріяхар.зменш.відносної щільності сечі до 1012-1006, при чому добові зміни незначні. Хар.пор.концентраційної здатності нирок; якщо на фоні олігурії – це ураж.і клубочків і канальців.
При повній втраті нирками здатності концентрувати сечу роз.ізостенурія, коли щільність сечі дор.щільності фільтрату 1010 та не змінюється проттягом доби (монотонний діурез).
110. Патологічні компоненти сечі: протеїнурія, циліндрурія, глюкозурія, аміноацидурія, гематурія, лейкоцитурія. Поняття про селективну і неселективну протеїнурію та її механізми.
Протеїнурія. Механізми: ушкодж.клуб.фільтра (клубочкова пр.), змен.реабсорбції (канальцева) та патол.надходження білка з туб.епітелію чи перитуб.лімф.рідини (секреторна). Є селективна та неселективна. Є нефротичний тип (альбуміни і або альфаглобуліни) та нефритичний тип (всі класи).
Гематурія обумовлена: 1)пошкодж.фільтру клубочків –виявляються вилужені Ер, 2)пошкодження сечовивідних шляхів.
Лейкоцитурія (>5 в п.з.) аж до піурії. Прчина – запалення нирк.тасечовивід.шл. Циліндрурія – зліпки ниркових канальців, які утв.при пошкодженні епітелію і склад.зі згорнутого білка (уропротеїнТамма-Хорсфалла) та мертвих клітин. Є гіалінові, зернисті та епітеліальні циліндри.
Глюкозурія. Пор.реабсорбції, позанирковогогенезу при всіх випадках гіперглікемії; ниркового генезу при хрон.хв.нирок, інток.свинцем, ртуттю, спадк.аномаліїдомін, мех- ураження ферментних систем (гексокіназа, Г-6-Фаза тощо).
Аміноацидурія – зб.АК в сечі, найчастіше при дефекті ферменту. Є Аргінінсукцинатнаацидурія, аспартатглутаматурія, триптофанурія, гомоцистинурія, кетоацидурія, фенілкетонурія та ін.
Клубочковапротеїнуріязумовленапорушеннямпроникностігломерулярногофільтра для білківплазми. При цьомузалежновідспіввідношеннябілковихфракційсечірозрізняютьселективну і неселективнупротеїнурію. Протеїнурію з високимумістомальбуміну та близькихйомуфракційбілканазиваютьселективною. В їїрозвиткупровідну роль відіграєвтратафункціональногоелектростатичногобар’єра. При неселективнійпротеїнурії в сечівизначаєтьсяальбумін, значнакількістьвисокомолекулярнихглобулінів, появуякихпов’язуютьпередусімізпорушеннямселек- тивності, залежноївідрозмірів пор гломерулярногобар’єра. Клубочковапротеїнуріяспостерігається при гломерулонефритах (ГН), нирковихваскулітах, діабетичнійнефропатії, амілоїдозінирок та іншихзахворюванняхнирок.
111. Етіологія та патогенез гострої ниркової недостатності. Клінічні прояви.
Етіологія:внутрішньониркові ф-ри (гострий гл-нефрит, пієлонефрит, тромбоз емболія, аренальна ГНД) та позаниркові ф-ри (шок, колапс, гемолітичні та міолітичні стани, зневоднення, екзо/ендогенна інтоксикація (солями важких Ме хлороформом токсикоз вагітних та діаб.кома), алергічні стани, при непрохідності сечовив.шляхів.
Патогенез 3 групи факторів: 1)пор.кровообігу в нирках (ішемія яка обум.гіповолемією, спазмом артеріол, ДВЗс, є звовротня ішемія (функціональна фаза ГНД) та незворотня (структурна фаза). 2)пряме ушкодження клубочків та канальців (нефротоксичні отрути, інф-ії; при ушкодж.прок. канальців зниж реабсорбція На –активація ренін –ангіотензинової с-ми – спазм артеріол – зменшення кровотоку та еф.фільтраційного тиску). 3)пор.відтоку сечі (при цьому зб.тиск у капсулі клубочків).
Клін. картина
Є 4 стадії: 1)початкова 2)олігоанурії (хар.гіперазотемія, пор.водно-ел.гомеостазу і КОС, клінічно набряк ГМ, та легень, зниження скоротливої ф-ї серця, екстрасистолії, брадикардії, блокади, дихання Куссмауля, гол.біль, блювота, арефлекія, судоми, кома, анемія.) 3)поліурії (5-10 діб, поновлення клубочкової фільтрації та зб.масифункц.нефронів, відновлення концентраційної ф-ї, амоніо та ацидогенезу тощо)4)одужання.
112. Сечовий та нефротичний синдроми: складові та механізми їх виникнення.
Сечовий синдром — клініко-лабораторний симптомокомплекс, пов’язаний з патологічними змінами показників сечі, не завжди специфічними скаргами, що відіграють важливу роль у діагностиці як ренальної, так і екстраренальної патології, дозволяє оцінювати активність захворювання та ефективність патогенетичної терапії.
Нефротичнийсиндром — клініко-лабораторнийсимптомокомплекс, пов’язаний із тривалою та значно підвищеною проникністю структур клубочків для білка, характеризується наявністю п’яти ознак: протеїнурії (понад 3 г/добу), гіпопротеїнемії за рахунок гіпоальбумінемії (у дорослих < 30 г/л, у дітей — < 25 г/л), диспротеїнемії за рахунок підвищення альфа-2-фракції глобулінів; набряків (від невиражених до анасарки); гіперліпідемії (гіперхолестеринемія понад 6,2 ммоль/л із можливою гіпертригліцеридемією). За відсутності набряків НС має назву неповного.
Патогенез
В основі НС лежить масивна протеїнурія — перший та провідний симптом цього патологічного стану. Вона виникає в результаті деструкції базальної мембрани клубочків (мембранозна нефропатія, що має неселективний характер) або малих відростків подоцитів (подоцитарна, селективна протеїнурія). Деструктивні зміни викликаються комплексом патологічних уражень із порушенням електростатичного бар’єра клубочків, до яких відносять:
— імунне запалення;
— відкладання амілоїду в базальній мембрані клубочка;
— активацію системи комплементу з наступними клітинними реакціями;
— значне підвищення вмісту К-залежного протеїну С, що впливає на V та VІІІ фактори згортання;
— локальне порушення згортання крові;
— токсичний вплив різних речовин на базальну мембрану клубочка та подоцити;
— підвищення судинної проникності;
— мікроангіопатії; — уроджена неповноцінність подоцитів;
— глікозилювання базальної мембрани клубочка при цукровому діабеті.
.
113.Етіологія та патогенез хронічної ниркової недостатності. Уремія. Уремічна кома
Етіологічними факторами хронічної недостатності нирок (ХНН) є хронічні прогресуючі захворювання нирок запальної (хронічний гломерулонефрит, хронічний пієлонефрит та ін.), судинної (гіпертонічна хвороба, стеноз ниркової артерії) та метаболічної (діабетичний гломерулосклероз, амілоїдоз, подагра) природи.
У патогенезі ХНН виділяють такі стадії: 1) початкову, 2) ранню поліуричну, 3) пізню олігуричну і 4) термінальну.
ХНН розвивається в результаті одночасного або поступового зменшення маси діючих нефронів (МДН).
Початкові ознаки ХНН з'являються при зменшенні МДН до 50-30 % від вихідної кількості нефронів, клінічно виражена картина розвивається при зниженні МДН до 30—10 % і величини клубочкової фільтрації нижче 20 % (якщо порівнювати з нормою). Дальше зменшення МДН і клубочкової фільтрації (нижче 10 %) веде до розвитку термінальної стадії недостатності нирок —уремії.
Уремічний синдром супроводжує розвиток гострої і хронічної недостатності нирок і розвивається внаслідок порушення клубочкової фільтрації. Перші його ознаки з'являються, коли швидкість клубочкової фільтрації (ІИКФ) стає нижче 50 мл/хв, а виражені клінічні симптоми - при падінні ШКФ нижче 10 мл/хв.
Основу уремічного синдрому становить інтоксикація, обумовлена накопиченням у крові кінцевих продуктів обміну речовин, які в нормі виводяться із сечею. Нині відомо понад 200 речовин, здатних викликати інтоксикацію при недостатності нирок. Найбільше значення мають:
а) сечовина;
б) сечова кислота;
в) похідні гуанідину — креатин, креатинін, метилгуанідин, диметилгуанідин та ін.;
г) ароматичні сполуки - феноли, індол, ароматичні аміни; ґ) аліфатичні аміни;
д) низькомолекулярні пептиди.
Ознакою накопичення зазначених токсичних речовин є азотемія - збільшення вмісту в крові залишкового азоту.
Клінічні прояви уремічного синдрому пов'язані з ураженням практично всіх органів і систем. Однак на перший план виступають ознаки токсичного ураження центральної нервової системи: анорексія, нудота, блювота, психічні порушення, ураження периферичних нервів.
Украй важким проявом інтоксикації є розвиток уремічної коми - повної втрати свідомості зі зникненням рефлексів на зовнішні і внутрішні подразники, пригніченням життєво важливих функцій. Крім інтоксикації, в патогенезі уремічного синдрому мають значення порушення обміну води, електролітів, розлади кислотно-основного стану.
114. Механізми розвитку загальних проявів при недостатності функції нирок: набряків, артеріальної гіпертензії, анемії, ацидозу, остеодистрофії.
Патогенетично розрізняють три типи набряків, що розвиваються при різних ураженнях нирок.
1. Набряки при гострій і хронічній недостатності нирок. Основний механізм їх розвитку — гідростатичний (гіперволемічний) (див. розд. 23). Зменшення швидкості клубочкової фільтрації, характерне для ниркової недостатності, призводить до затримки натрію й води в організмі (позитивний водний баланс) і, як наслідок, до гіперволемії. Остання, будучи причиною збільшення гідростатичного тиску в капілярах, викликає розвиток набряків за механізмом Старлінга.
2. Нефротичні набряки. Основний механізм їх розвитку — онкотичний (гіпопроте-гнемічний). Порушення клубочкового фільтра при нефрозі викликають масивну протеїнурію, у результаті якої розвивається гіпопротеїнемія і падає онкотичний тиск крові. Це, у свою чергу, за механізмом Старлінга викликає перехід води з судин у тканини - розвиваються набряки.
3. Нефритичні набряки. Розвиваються при гострому і хронічному гломерулонефриті. Патогенез цих набряків складний і в своїй основі має такі механізми:
а) запалення клубочків → застій крові в судинах нирок → гіпоксія юкстагло-мерулярного апарату → активація ренін-ангіотензинної системи → секреція альдостерону → затримка натрію в організмі і підвищення осмотичного тиску крові → секреція антидіуретичного гормону → затримка води → гі-перволемія → набряки;
б) запалення клубочків → порушення ниркового кровообігу → зменшення швидкості клубочкової фільтрації → затримка натрію й води в організмі → гіперволемія → набряки;
в) запалення клубочків → збільшення проникності ниркового фільтра →про-теїнурія → гіпопротеїнемія → набряки.
Які порушення КИСЛОТНО-ОСНОВНОГО стану можуть розвиватися при ураженнях нирок?
Найчастіше розвивається негазовий ацидоз. Залежно від сутності патологічних процесів у нирках можливі три його варіанти.
1. Клубочковий ацидоз (нирковий азотемічний ацидоз). Виникає внаслідок розвитку ниркової недостатності при зменшенні швидкості клубочкової фільтрації ниж-
че 25 мл/хв. Він є метаболічним ацидозом зі збільшеною аніонною різницею (див. розд. 25). Його розвиток обумовлений накопиченням іонів водню, що утворюються ендогенно, в основному у формі сірчаної, фосфорної, сечової та інших кислот.
2. Проксимальний нирковий канальцевий ацидоз. Є наслідком первинних порушень реабсорбції гідрокарбонату в проксимальних звивистих канальцях не-фронів. Його відносять до видільних ацидозів з нормальною аніонною різницею (гіперхлоремічний).
3. Дистальний нирковий канальцевий ацидоз. Обумовлений первинними порушеннями процесів ацидогенезу в дистальних звивистих канальцях, де відбувається відтитровування буферів (збереження гідрокарбонату) і підкислення (аци-дифікація) сечі. Є метаболічним ацидозом з нормальною аніонною різницею (гіперхлоремічний). Виявляється нездатністю витримувати ендогенне навантаження іонами водню. При цьому рН сечі не досягає значень нижче 6.
Зміни функції нирок іноді можуть спричинятися до розвитку негазового гіпо-каліємічного алкалозу. Це, зокрема, буває при гіперальдостеронізмі. Альдостерон, посилюючи секрецію іонів калію в дистальних звивистих канальцях, викликає гіпо-каліємію, яка, у свою чергу, обумовлює виникнення алкалозу.
Які порушення фосфорно-кальцієвого обміну характерні для недостатності нирок? Який їх патогенез?
Порушення фосфорно-кальцієвого обміну при недостатності нирок виявляються розвитком ниркової остеодистрофії (остеопатії).
Ниркова остеодистрофія — це комплекс дистрофічних порушень у кістках, що виникають унаслідок розладів фосфорно-кальцієвого обміну при ураженнях нирок. Клінічно вона виявляє себе двома групами змін.
I. Дистрофічні зміни в кістках:
а) резорбція кісткової тканини (фіброзно-кістозний остеїт) — у кістках утворюються порожнини, які заповнюються фіброзною тканиною. Є наслідком гіперпаратиреозу;
б) остеомаляція - розм'якшення кісток, їх деформація, болі в кістках. У дітей порушується окостеніння хрящів. Є наслідком гіпокальціємії;
в) остеопороз — зменшення щільності кісткової тканини без деформації кісток;
г) остеосклероз — збільшення щільності кісткової тканини.
II. Обвапнеїшя м'яких тканин - кальцифікація (див. розд. 24).
У патогенезі ниркової остеодистрофії мають значення такі механізми:
а) хронічна недостатність нирок (ХНН) → зменшення екскреції фосфатів → гіперфосфатемія → гіпокальціємія (див. розд. 24) → збільшення продукції паратирину → резорбція кісткової тканини, її демінералізація;
б) ХНН → порушення перетворення 25-оксивітаміну D у його гормонально активну форму- 1,25-діоксивітамін D → зменшення всмоктування іонів Са2+ у кишках → гіпокальціємія → див. вище;
в) ХНН → клубочковий ацидоз → обмін іонів Са2+ і Na+ на іони Н+ крові (буферна функція кісткової тканини) → демінералізація кісток.
Які види артеріальної гіпертензії можуть виникати при ураженнях нирок? Який їх патогенез?
Артеріальна гіпертензія є одним з проявів хронічної недостатності нирок (ХНН). Виділяють такі її види.
1. Гіпертензія, залежна від об'єму. Цей вид буває у 80-90 % хворих з ХНН. Гї патогенез пояснюють таким чином: ХНН → зменшення швидкості клубочкової фільтрації → порушення екскреції натрію і води → гіперволемія → збільшення хвилинного об'єму серця → збільшення артеріального тиску.
2. Гіпертензія з високим рівнем реніну (високоренінова). Цей вид виявляють в.І 5-10 % хворих з ХНН. Основний її механізм: ураження нирок → порушення ниркового кровообігу → гіпоксія юкстагломерулярного апарату → вивільнення реніну → активація ренін-ангіотензинної системи → далі див. розд. 28.
3. Гіпертензія, не залежна від об'єму. Буває в 5-10 % хворих з ХНН.
Характеризується нормальним об'ємом циркулюючої крові й нормальним рівнем реніну в крові. Вважають, що розвиток цього виду гіпертензії пов'язаний з порушенням депресорних функцій нирок, а саме — зі зменшенням утворення ниркових простагландинів, зокрема ПГ Е2, зменшенням активності ниркової калікреїн-кінінової системи, пригніченням утворення нейтрального ліпіду, що має гіпотензивну дію.
Які механізми обумовлюють розвиток анемії, що супроводжує недостатність нирок?
Ознаки анемії у хворих з хронічною недостатністю нирок з'являються при зменшенні швидкості клубочкової фільтрації нижче 40 мл/хв. У її патогенезі мають значення такі механізми.
1. Пригнічення еритропоезу:
а) порушення регуляції еритропоезу, обумовлене пригніченням утворення ниркових еритропоетинів і посиленим продукуванням інгібіторів кровотворення;
б) ушкодження кровотворних клітин уремічними токсинами;
в) дефіцит заліза, що розвивається як наслідок хронічних крововтрат при захворюваннях нирок і втрат трансферину в результаті протеїнурії.
2. Крововтрати. Можуть бути пов'язані з гематурією, з частим утворенням виразок у шлунку і кишках хворих з ураженням нирок, з розвитком геморагічного діатезу, і
3. Гемоліз еритроцитів, обумовлений уремічними токсинами.
115. Гострий та хронічний дифузний гломерулонефрит: етіологія, експериментальні моделі, патогенез.
Гломерулонефрит - це двостороннє дифузне захворювання нирок алергічної природи.
Розрізняють гострий і хронічний гломерулонефрит.
Для гострого гломерулонефриту характерні бурхливий початок, олігурія, протеїнурія, азотемія, артеріальна гіпертензія, набряки, гематурія, порушення з боку центральної нервової системи.
Гострий гломерулонефрит виникає під час (або після) якої-небудь інфекції, частіше стрептококової природи. Вважають, що гемолітичний стрептокок групи А (типи 4,12) є специфічним "нефритогенним" штамом. Певну роль відіграють й інші інфекції, у тому числі вірусні, паразитарні. Встановлено етіологічну роль охолодження, дифузних уражень сполучної тканини (системний червоний вовчак, ревматоїдний артрит, вузликовий періартеріїт), опікової хвороби, попередньої вакцинації або використання з лікувальною метою гетерологічних сироваток.
Виділяють два основних патогенетичних варіанти гострого гломерулонефриту.
1. Ураження базальної мембрани клубочків нефронів антитілами проти її антигенів - нефротоксичний гломерулонефрит (має стрімкий перебіг, швидко прогресує). Носієм антигенних властивостей базальної мембрани є глікопротеїд.
2. Розвиток запального процесу в клубочках унаслідок фіксації на базальній мембрані та всередині її імунних комплексів - імунокомплексний гломерулонефрит.
У розвитку такого типу хвороби можуть брати участь антигени як екзогенного (інфекційної чи неінфекційної природи), так і ендогенного (білок тканин, ДНК) походження. Антитіла (Ig G, Ig М) прямо в крові вступають у взаємодію із зазначени-
ми антигенами, потім у вигляді імунних комплексів (антиген-антитіло-комплемент) надходять у клубочки, відкладаючись на їхній базальній мембрані. Реалізація ушкоджувальної дії імунних комплексів, як і нефротоксичних антитіл, відбувається через розвиток імунного запалення (див. розд. 10).
До імунокомплексних відносять гломерулонефрити, що розвиваються після стрептококової інфекції, при системному червоному вовчаку, сироватковій хворобі та ін. Більшість випадків гломерулонефриту (не менше ніж 80 %) є імунокомплексними.
Хронічний гломерулонефрит являє собою тривале прогресуюче дифузне двостороннє ураження нирок запальної природи, неоднорідне за походженням, клінічними проявами і патогенезом.
Залежно від клінічних проявів виділяють такі його форми.
1. Латентна форма виявляє себе ізольованим сечовим синдромом — помірною про-теїнурією, гематурією. У частини хворих спостерігають набряки і транзиторну артеріальну гіпертензію.
2. Гіпертонічна форма характеризується стійким підвищенням артеріального тиску, набряками, гематурією, протеїнурією, циліндрурією і лейкоцитурією.
3. Нефротична форма, яку супроводжують набряковий синдром, виражена протеї-нурія і циліндрурія, зміни крові (гіпопротеїнемія, гіперліпідемія).
4. Змішана, або нефротично-гіпертонічна форма, для якої характерні набряк і гіпертензія, але на відміну від нефротичної форми нема характерних змін крові.
Хронічний гломерулонефрит може бути наслідок гострого, але частіше розвивається первинно. Залежно від причин виділяють такі його форми:
1) інфекційного походження (постстрептококовий, при затяжному септичному ендокардиті, малярії, сифілісі, туберкульозі та інших інфекціях);
2) неінфекційні (сироватковий, вакцинний, медикаментозний, травматичний, при отруєннях, охолодженні, тромбозі ниркових вен);
3) зумовлені дифузними захворюваннями сполучної тканини (ревматоїдний артрит, системний червоний вовчак, геморагічний васкуліт та ін.);
4) особливі (після еклампсії, радіаційний, спадковий та ін.).
Загальновизнаною є імунологічна концепція розвитку хронічного гломерулонефриту. Поряд з нефротоксичними та імунокомплексними механізмами (див. запит. 32.29) певне значення в його патогенезі має гіперчутливість уповільненого типу.
Які існують експериментальні моделі гострого гломерулонефриту?
1. Введення тваринам нефротоксичної сироватки, тобто сироватки, що містить антитіла проти антигенів ниркової тканини.
Серед моделей цієї групи найчастіше використовують модель Ліндемана і модель Масугі.
Модель Ліндемана. Кролям внутрішньовенно вводять нефротоксичну сироватку, отриману від морської свинки, попередньо імунізованої суспензією нирки кроля. При цьому відбувається фіксація протиниркових антитіл на базальних мембранах клубочків з наступним зв'язуванням комплементу і розвитком ушкодження. Модель Масугі. Кролям внутрішньовенно вводять нефротоксичну сироватку качок, імунізованих тканиною нирок кролів. Особливість цієї моделі полягає в тому, що антитіла птахів фіксуються на базальній мембрані клубочків, але не можуть зв'язувати комплемент ссавців. Це призводить до утворення власних кролячих антитіл проти фіксованих качиних (для цього треба 6—8 діб). Останні в цьому випадку виступають антигенами. Кролячі антитіла, з одного боку, взаємодіють з фіксованими на базальній мембрані качиними, з другого - зв'язують комплемент,
активація якого і призводить до ушкодження клубочків.
2. Охолодження нирки — заморожування хлороформом (Герцен). При цьому в крові дослідних тварин (кролів) з'являються специфічні протиниркові антитіла і зазнає уражень друга інтактна нирка. Зазначена модель доводить роль аутоімунних механізмів у розвитку так званого "окопного" нефриту (нефриту воєнного часу).
3. Введення в черевну порожнину кролів клітинної суспензії ниркової тканини і культури стрептококів (подружжя Ковелті).
4. Імунізація овець базальними мембранами клубочків нефронів нирки людини в повному ад'юванті Фрейнда (Стеблей).
5. Імунізація щурів суспензією гомологічної або аутологічної нирки з повним ад'ю-вантом Фрейнда (Хеймен).
6. Виведення чистих ліній тварин (наприклад, новозеландські миші лінії NZB/BL), у яких спонтанно виникають аутоімунні хвороби, у тому числі і гломерулонефрит.
116. Особливості розвитку гострого гломерулонефрита у дитячому віці: причини, патогенез та прояви.
Див. вище
117. Етіопатогенез гемолітико-уремічного синдрому (ГУС) у дітей.
Гемолітико-уремічний синдром (ГУС) – патологічний синдром, для якого характерна тріада: поєднання гострої мікроангіопатичної гемолітичної анемії, тромбоцитопенії, гострої ниркової недостатності. Залежно від етіології виділяють дві основні форми ГУС: 1) стан, що виникає після перенесеної гострої діареї (Причиною є вплив Шига-токсину Shigella dysenteriae), 2) ГУС, що розвинувся без попереднього епізоду діарейного захворювання. (Причинами бути як генетичні фактори, такі як дефіцит фактора Н комплементу, дефіцит антитромбіну ІІІ, порушення синтезу простациклінів, порушення обміну кобаламіну та інші, так і інфекції – пневмококова, анаеробна, ентеровірусна, грипозна, герпетична, ВІЛ; на фоні застосування деяких лікарських препаратів . Патогенез ГУС : під дією Шига-токсину штамів шигел ( серогрупи А та ЕГЕК) відбувається ураження ендотелію судин - ураження токсином рибосом, що призводить до порушення синтезу білка і подальшої загибелі клітин. Розвиток ГУС супроводжується активацією системного запалення з розвитком синдрому системної запальної відповіді (ССЗВ). Максимальна вираженість ураження ендотелію судин відбувається в клубочках нефронів. Ураження ендотелію судин призводить до активації системи зсідання крові й розвитку тромбозів судин мікроциркуляторного
русла. відбувається поглинання тромбоцитів -> розвиток тромбоцитопенії.
Ураження клубочків та канальців нирок призводить до розвитку гострої ниркової недостатності (ГНН). Анемія виникає внаслідок руйнування еритроцитів при проходженні крізьзмінені внаслідок відкладення фібрину капіляри, фрагментація еритроцитів відбувається під дією цитокінів, які виділяються нейтрофілами.
118.Етіопатогенез синдрому Фанконі у дітей.
Етіологія
Синдром Фанконі може бути:
спадковим
захворювання, як правило, супроводжує інші генетичні порушення, зокрема, цистиноз. Крім дисфункції ниркових канальців, розвиваються інші ускладнення цістіноза: розлади зору, гепатомегалія, гіпотиреоз та інші клінічні прояви. Синдром Фанконі також може супроводжувати хвороба Вільсона, спадкову непереносимість фруктози, галактоземії, окулоцереброренальний синдром (синдром Лоу), мітохондріальні цітопатіі і Тирозинемія.
2) придбаним
Це порушення може бути викликано різними лікарськими препаратами, включаючи хіміотерапію при раку (наприклад, ифосфамид, стрептозоціна), антиретровірусні препарати (наприклад, диданозин, цидофовир) і застарілий тетрациклін. Набутий синдром Фанконі також може розвиватися після трансплантації нирки і у пацієнтів з множинною мієломою, амілоїдозом, інтоксикацією важкими металами або іншими хімічними речовинами, або при дефіциті вітаміну D.
Основна ланка патогенезу — мітохондріальний ферментний дефект в циклі Кребса, ферментна тубулопатія, що характеризується порушенням реабсорбції глюкози, амінокислот, фосфатів і бікарбонатів в канальцях нирок. Втрата амінокислот і бікарбонату сприяє розвитку метаболічного ацидозу - > посилюється резорбція кісткової тканини і знижується реабсорбція калію і кальцію в канальцях нирок -> розвиток гіпокаліємії і гіперкальціурії. Втрата фосфору веде до розвитку рахіту. мітохондріальний ферментний дефект в циклі Кребса веде до порушення процесів енергозабезпечення, реабсорбції фосфатів , глюкози і амінокислот в ниркових канальцях і підвищеної їх екскреції з сечею — порушується кислотно — основний стан, а метаболічний ацидоз і брак фосфатів сприяють руйнуванню кісткової тканини по типу рахітоподібних змін скелета і остеомаляції .
119.Загальні механізми порушення функції ендокринної системи (гіпофункція, гіперфункція, дисфункція ендокринних залоз; первинні, вторинні ендокринопатії).
Патогенетичні механізми порушень ендокринної системи
I. Дисрегуляторні порушення - розлади регуляції ендокринних залоз.
II. Залозисті порушення — розлади біосинтезу гормонів та їх секреції.
III. Периферичні порушення — розлади транспорту, рецепції і метаболізму
У разі первинного ушкодження залози внутрішньої секреції розвивається первинна ендокринна паталогія, у випадках коли органічні ураження або функціональні розлади спочатку виникають за її межами — вторинна. Найчастіше первинна ендокринна патологія розвивається як автоімунний процес наприклад аутоімунний тириоїдит, пухлина - аденома гіпофіза, некроз або крововилив, вроджені вади біосинтезу гормонів. У виникненні вторинної ендокринної патології переважне значення мають розлади нейрогормональної регуляції секреції гормонів наприклад псевдоспіввідношення продукції лютропіну і фолітропіну при синдромі полікістозних яєчників. До групи вторинних гормональних порушень належать і периферичні.
ТИПИ порушень ендокринних функцій
I. Гіперфункція ендокринних залоз (ендокринна гіперфункція).
II. Гіпофункція ендокринних залоз (ендокринна гіпофункція).
III. Дисфункція ендокринних залоз (ендокринна дисфункція) — різноспрямовані зміни функції ендокринних залоз.
Основою патогенезу більшості ендокринних хвороб є недостатня — гіпофункція, або надмірна — гіперфункція, активність ендокринних залоз і клітин. Будь-яка залоза є джерелом 2 або більше кількості гормонів. У випадку ураження залози внаслідок крововиливу або запального процесу припиняється або ослаблюється продукція гормонів. Наприклад некроз аденогіпофіза призводить до генералізованної аденогіпофізарної недостатності. Ізольоване порушення секреції того чи іншого гормону — парціальна ендокринна гіпер або гіпо функція (хвороба Іценка — Кушинга — надмірна секреція кортикотропіну.)
Дисфункція зумовлена різноспрямованими змінами синтезу гормонів і близьких до них фізіологічно активних сполук в одному і тому самому ендокринному органі або ж утворенням і надходженням в кров аномальних за молекулярною будовою і біологічними властивостями гормональних речовин. Патогенез уродженоюго адреногенітального синдрому зумовлює появу жіночого несправжнього гермафродитизму, що пов’язаний із блокадою певних ферментів біосинтезу стероїдів у кірковій речовині наднирників. Це зумовлює різке зменшення утворення кортизолу з подальшим збільшенням продукції надниркових андрогенів.
120.Поняття ендокринної регуляції. Дисрегуляторні ендокринопатії.
Ендокринна регуляція або ендокринна функція — участь ендокринних залоз у здійсненні гуморальної регуляції структурних змін, фізіологічних і біохімічних процесів в організмі. Має такі складові частини: регуляція діяльності ендокринних залоз, власне функція ендокринних залоз — утворення і секреція гормонів, транспорт гормонів до клітин — мішеней, Взаємодія гормонів з периферичними клітинами, екскреція гормонів.
Дисрегуляторні ендокринопатії. В основі развитку лежать розлади регуляції эндокринних залоз, а саме 4 механизмів : 1) нервовий – Прямий нервовий вплив регулює діяльність мозкового шару наднирників, гіпоталамуса, епіфіза. 2) нейроендокринний — секреція Releasing — гормонів що регулюють діяльність аденогіпофіза. 3) Ендокринний — безпосередній вплив одних гормонів на синтез і секрецію інших — дія тропних гормонів аденогіпофіза на кору наднирників, статеві залози. 4) неендокринно гуморальний — здійснюється неспецифічними гуморальнами факторами, метаболітами та іонами. Концентрація глюкози в крові прямо впливає на синтез секрецію інсуліну та глюкагону. Розвиток дисрегуляторних ендокринопатій може бути пов’язаний з розладами всіх механізмів регуляції. В 1 випадках розвивається ендокринна гіпер функція в інших при пригнічені регуляторних впливів — ендокринна гіпо функція.
В основі регуляції ендокринної функції лежать 2 основних принципи: прямих зв’язків і зворотних зв’язків.
Порушення негативних зворотніх зв’язків. Приклад. Система гіпоталамус — аденогіпофіз — щитоподібна залоза. Дефіцит йоду в продуктах харчування і питній воді —> зменшення утворення тиреоїдних гормонів —> стимуляція гіпоталамуса і аденогіпофіза—> збільшення утворення тиреоліберину і тиреотропного гормону — >гіпертрофія щитоподібної залозии —> ендемічний зоб
121. Залозисті ендокринопатії: причини, механізми та прояви.
Залозисті ендокринопатії обумовлені: 1)зміною к-сті ендокринних клітин (із відповідно гіпер (пухлина) та гіпо (ураження чи видалення) функцією) 2 )якісними змінами ендокриноцитів (порушення біосинтезу чи секреції). Порушення біосинтезу: 1)білково-пептидних гормонів (порушення транскрипції, трансляції, дефіцит АТФ, порушення посттрансляційної модифікації). 2)стероїдних г. (пор.обміну холестерину, ферментопатії, гіпоксія чи дефіцит НАДФ –для гідроксилювання, дефіцит вихідних Амінокислот, деф.мікроелементів, ферментопатії, деф.АТФ).Порушення секреції: 1)порушення депонування гормонів. (пор.утв.комплексів, напр. білки-нейрофізини для вазопресину та окситоцину, цинк для інсуліну тощо). 2)порушення передачі сигналів, які стимулюють секрецію (пор.утв. цАМФ, Са) 3)ураження контрактильних елементів (мікрофіламентів/трубочок, які потрібні для екзо/ендоцитозу гормонів) 4)дефіцит АТФ.
122. Периферичні ендокринопатії: причини, механізм та прояви.
Причини і механізми :
Порушення транспорту гормонів в організмі
Гормон може транспортуватись 4 шляхами: вільно (розчиненим у воді), в комплексі зі специфічними транспортними чи неспецифічними білками плазми і адсорбуючись на поверхні формених елементів крові. При збільшенні звязування гормону зменшується вміст його вільної форми → ендокринна гіпофункція. При зменшенні звязування, збільшується концентрація вільної форми → ендокринна гіпофункція. Розлади метаболічної інактивації гормонів
При уповільненні інактивації гормонів збільшується їх вміст у крові → ендокринна гіперфункція, і навпаки.
Порушення взаємодії гормонів з периферичними клітинамимішенями – патологія циторецепції гормонів Для гормонів характерні різні типи взаємодії з клітинами: внутрішньоклітинний (тиреоїдні і стероїдні гормони) і мембранний (білково-пептидні гормони і катехоламіни) типи. Також існує 2 варіанти патології клітинних рецепторів: зменшення кількості рецепторів або їхньої спорідненості до гормону (десенситизація) та збільшення кількості рецепторів до гормону (сенситизація).
Проявами кожного з цих механізмів розвитку периферичної ендокринопатії може бути ендокринна гіперфункція або ендокринна гіпофункція.
123. Патологія гіпоталамо-гіпофізарної системи. Причини виникнення та механізми розвитку синдромів надлишку та нестачі гіпофізарних гормонів.
Причини порушення діяльності гіпоталамо-гіпофізарної системи:
Патогенна дія факторів зовнішнього і внутрішнього середовища
(негативні емоції, біль, психічні порушення)
Ураження регуляторних відділів ЦНС – вищих центрів кори великих півкуль, структур лімбічної системи, ретикулярної формації.
Ураження гіпоталамуса
Ураження гіпофіза
Аденогіпофіз синтезує: СТГ, АКТГ, ТТГ, ФСГ, ЛГ та пролактин. При недостатності гормонів аденогіпофіза порушується діяльність периферичних ендокринних залоз (щитоподібної, статевих, кори надниркових залоз).
Надлишок гіпофізарних гормонів утворюється при доброякісних пухлинах (аденоми) чи гіперплазії аденогіпофізу. Клінічно це проявляється так: При наявності еозинофільних аденом гіпофіза(пролактинома, аденома соматотрофів):
Синдром гіперпродукції пролактину (припинення менструацій, витікання молока, безпліддя, відсутність лібідо)
При гіперсекреції СТГ виникає гігантизм та акромегалія, крім того, гіперглікемія, інсулінорезистентність, метагіпофізарний цукровий діабет.
При наявності базофільних аденом гіпофіза (ростуть із кортикотрофів, тиротрофів, гонадотрофів), найчастіше кортикотрофна, вона викликає хворобу Іценка-Кушинга, яка проявляється:
Вторинний гіперкортицизм (↑ утворення кортизолу)
Посилена пігментація шкіри
124.Етіологія, патогенез, клінічні прояви пангіпопітуітаризму. Причини, механізми, клінічні прояви парціальної недостатності гормонів аденогіпофіза (СТГ, ТТГ, АКТГ, гонадотропінів).
Пангіпопітуїтаризм – це повна недостатність гіпофіза.
Етіологія. Недостатність гіпофізу буває вродженою і набутою. До причин хвороби найчастіше належать:
Пухлина (хромофобна аденома гіпофіза)
Некроз гіпофіза після пологів – синдром Шихана
Травма основи черепа
Запалення
Тромбоз
Вірусна інфекція
Іонізуюча радіація та променева терапія
Патогенез. Більшість порушень повязана з припиненням синтезу СТГ, ТТГ, АКТГ.
Клінічні прояви: млявість, малорухливість, зниження основного обміну, схильність до гіпоглікемії, гіпотермія, зниження мязового тонусу і резистентності до несприятливих впливів. Ураження залози в ембріональний період або перед статевим дозріванням призводить до карликовості, статевого недорозвинення, зниження функції щитоподібної залози, ендокринно-обмінних порушень, зниження загальної реактивності.
При руйнуванні 95% залози – розвивається гіпофізарна кахексія (хвороба Сіммондса), за якої визначається загальне виснаження, атрофія щитоподібної, надниркових і статевих залоз, мязів, внутрішніх органів, руйнування кісток, волосся, зубів, гіпоглікемія, розлад нервової системи. Парціальна (часткова) недостатність гормонів аденогіпофіза має ту ж етіологію, що і пангіпопітуітаризм, та спричиняє
Любимый муж, [07.06.20 18:43]
зниження рівня якогось одного гормону аденогіпофіза.
Клінічні прояви:
При ранній недостатності СТГ – гіпофізарна карликовість, гіпогонадизм
При гонадотропній недостатності - вторинний гіпогонадизм, фзіичне недорозвинення, затримка статевого розвитку – гіпофізарний інфантилізм (затримка менструацій, безплідність, затримка опущення яєчок в калитку(крипторхізм)).
При недостатності ТТГ – вторинний гіпотиреоз.
При недостатності АКТГ – вторинний гіпокортицизм.
125.Причини та прояви гігантизму та карликового нанізму у дитячому віці. Принципи патогенетичної корекції гормональних порушень
Гіпофізарний гігантизм – нейроендокринне захворювання, викликане хронічною надлишковою секрецією ГР у дітей та підлітків, характеризується пропорційним ростом кісток скелету в довжину, що призводить до значного збільшення лінійного росту хворого.Причиною захворювання є еозінофільна аденома передньої частки гіпофізу (95%), у деяких випадках – пухлина гіпоталамусу. Гіперфункцію еозинофільних клітин можуть спричинити травма черепа, інфекційні та інші запальні процеси в проміжно-гіпофізарній ділянці. Патологічно високий зріст може бути зумовлений підвищеною чутливістю епіфізарних хрящів до гормону росту. В 90% випадків причиною гігантизму є макроаденома, а у 9% – мікроаденома гіпофізу. Вкрай рідко гігантизм виникає на тлі синдрому «порожнього» турецького сідла.
Гігантизм у дітей проявляється, як правило, під час статевого дозрівання. У виняткових випадках діти-гіганти народжуються у батьків з цією патологією. Хлопці страждають на гігантизм у 9 разів частіше, ніж дівчата.
Клініка захворювання характеризується надмірним, відносно пропорційним ростом і дещо подовженими кінцівками. Згодом у гіпофізарних гігантів можуть розвиватися акромегалоїдні риси (акромегалічний гігантизм). У 40% гігантів відзначається акромегалія, у 20% акромегалів – гігантизм.
На перший план виступає високий зріст, грубі риси обличчя на тлі загального інфантилізму
У гіпофізарного гіганта спостерігається м’язова слабкість, підвищена втомлюваність, головокружіння, запаморочення, головний біль, біль у кінцівках, погіршення пам’яті, серцебиття. Тембр голосу стає низьким. Шкіра – масляниста, волога. Риси обличчя у підлітка грубіють, спостерігається прогнатизм, виражені шкірні зморшки, гіпертрихоз, акантоз, фіброзні шкірні поліпи, гіперпігментації.
Пізніше, майже у всіх хворих, з’являються зміни з боку скелету, розвивається кіфоз або кіфосколіоз, нижні кінцівки набувають Х-подібної форми. У дітей раннього віку швидко ростуть довгі кістки. На рентгенограмі виявляють стовщення кісток у місці прикріплення м’язів, стовщення кісток черепа, посилення пневматизації основної та придаткових пазух, подовження і звуження п’ясткових кісток і фаланг
Гіпофізарний нанізм розвивається внаслідок первинної недостатності гормону росту (ГР, соматотропіну, СТГ) та характеризується пропорційною затримкою росту і розвитку дитини.
Дефіцит гормону росту (гіпосоматотропінемія) може бути вродженим або набутим, обумовленим різними причинами.
Розрізняють також органічну (результат внутрішньочерепного ушкодження) та ідіопатичну форму (при відсутності специфічної органічної патології гіпоталамо-гіпофізарної області) дефіциту ГР. Найчастіше визначається ідіопатична форма соматотропної недостатності
Основними клінічними ознаками недостатності гормону росту є:
- маса тіла при народженні зазвичай нормальна;
- дуже погане збільшення в рості з раннього дитинства;
- надлишкова маса тіла;
- інфантильна зовнішність;
- статеве недорозвинення;
- виражене відставання у рості;
- низька швидкість росту;
- затримка дозрівання кісткової тканини;
- нормальні пропорції тіла.
Немовлята, які мають недостатність СТГ, не відрізняються від здорових дітей, оскільки внутрішньоутробний дефіцит гормону росту не призводить до затримки росту. Відставання в рості зазвичай виявляється до 5-річного віку.
Діти ростуть дуже повільно. На тлі різкого відставання у рості, затримці дозрівання кісток, зниженої швидкості росту в дитини зберігаються нормальні пропорції тіла. Часто відзначається надлишок маси тіла (частіше – на тулубі, при цьому відкладення жиру рівномірне). Шкіра суха, часто – з жовтим відтінком, волосся тонке, ламке, голос – високий. В лобно-скроневій ділянці – рідке тонке волосся.
Скелет та внутрішні органи зменшені у розмірах (спланхомікрія), кістки обличчя недорозвинуті, перенісся западає («лялькове обличчя»). Відносно великий лоб може свідчити про вроджену гідроцефалію.
Статевий розвиток різко загальмований. Кістковий вік, як правило, складає 1/2 від паспортного, «зони росту» у таких хворих залишаються відкритими навіть в дорослому віці. Значна частина пацієнтів має супутній дефіцит ТТГ, гонадотропінів, рідше – АКТГ.
126. Патофізіологія нейрогіпофізу. Нецукровий діабет: причини, механізми та прояви. Принципи патогенетичної корекції гормональних порушень.
Патофізіологія нейрогіпофізу проявляється патологічним синтезом вазопресину і окситоцину: їх надлишком, або недостатністю.
Надлишок вазопресину – синдром неадекватної секреції вазопресину (синдром Пархона): причиною може бути:
Пухлини різних тканин, що продукують вазопресин
порушена інактивація гормону в печінці при цирозі
больовий шок
розлади регуляції ендокринної функції гіпоталамуса
Прояви: сильний антидіуретичний ефект → накопичення води в організмі → рефлекторна анурія → підвищення АТ, ↓ Na в крові (вторинна гіпонатріємія), ↓осмолярність сироватки крові →внутрішньоклітинний набряк, набряк головного мозку, порушення ЦНС, але ОЦК залишається сталим, тому набряків периферичних тканин немає.
Дефіцит вазопресину Причини:
Абсолютний дефіцит- ураження ділянок нейроендокринної системи (супраоптичного і паравентрикулярних ядер гіпоталамуса)
Відносний дефіцит - підвищена інактивація гормону або недостатня чутливість до вазопресину дистальних частин канальців нефронів. Прояви: нецукровий діабет, що має 2 патогенетичних варіанти: центральний (при абсолютному дефіциті вазопресину) і нефрогенний (при відносному дефіциті).
Абсолютний або відносний дефіцит вазопресину в крові → порушення реабсорбції води в канальцях нефронів → поліурія (3-8 л сечі за добу, іноді до 25-40 л)→ ↓ОЦК → спрага (полідипсія), ↓АТ, гіпоксія.
Дефіцит окситоцину викликає порушення пологової діяльності, лактації, дискінезія жовчних шляхів.
127. Недостатність кори наднирників: види (первинна, вторинна; гостра, хронічна). Етіологія, патогенез, клінічні прояви хронічної наднирникової недостатності. Хвороба Аддісона.
I. За етіологією розрізняють первинну і вторинну недостатність кори надниркових залоз. Первинна виникає як результат власне ураження надниркових залоз, вторинна пов'язана або з ураженнями гіпоталамуса (дефіцит кортиколіберину), або з гіпофункцією аденогіпофіза (дефіцит АКТГ).
II. Залежно від характеру порушень виділяють тотальну (порушено утворення всіх гормонів) і парціальну (як правило, спадково обумовлені дефекти синтезу окремих гормонів) недостатність кори надниркових залоз.
III. За клін. перебігом недостатність кори надниркових залоз може бути гострою і хронічною.
Прикладами гострої недостатності є:
а) стан після адреналектомії (видалення надниркових залоз);
б) крововиливи в надниркові залози, що виникають при сепсисі, особливо при менінгококовій інфекції (синдром Уотерхауза—Фредріксена);
в) синдром відміни глюкокортикоїдних препаратів.
Хронічна недостатність кори надниркових залоз відома під назвою хвороби Аддісона (бронзовоїхвороби).
Найчастіші її причини:
а) туберкульозне руйнування надниркових залоз;
б) аутоімунне їх ушкодження.
Хронічна недостатність кори надниркових залоз у людини відома під назвою хвороби Аддісона, або бронзової хвороби. Вона виникає найчастіше при туберкульозі надниркових залоз, а також атрофії кіркової речовини після перенесених важких інфекц. захв-нь або тривалого лікування кортикостероїдними препаратами.
Для хронічного гіпоадренокортицизма характерні схуднення, швидка фізична і психічна стомлюваність, поганий апетит, дисфункція травного каналу, артеріальна гіпотензія, прогресуюча гіперпігментація шкіри.
Механізм гіперпігментації пов'язаний з посилення крейд-цит стимулюючої активності гіпофіза, яка супроводжує виникнення при гіпоадренокортицизмі збільшення секреції кортикотропіну. Різноманітні патогенні впливи - травма, інфекція, крововтрата і навіть екстракція зуба - у хворого на аддісонову хворобу можуть привести до гострої недостатності кори надниркових залоз.
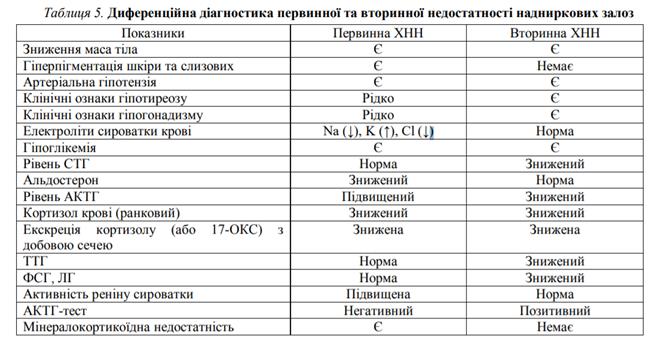
128. Синдром відміни глюкокортикоїдів. Петля зворотного зв’язку у ендокринній регуляції, принципи призначення та відміни глюкокортикоїдів.
Синдром відміни. За тривалої глюкокортикоїдної терапії може розвинутися характерний синдром відміни.
Для легкої його форми (стероїдний псевдоревматизм) характерні: артралгія, міалгія, диспептичні явища, емоційна лабільність, головний біль, депресія, нездужання, безсоння, лущення шкіри. Ці прояви швидко купіруються при призначенні адекватних доз глюкокортикоїдних препаратів.
При тяжких формах синдрому відміни розвивається панмезенхімальна реакція з гарячковим станом і генералізацією запальних реакцій (включаючи серозити та легеневі інфільтрати), що можуть трансформуватися в панартеріїт та системний червоний вовчак. Іноді синдром відміни нагадує клін. прояви основного захворювання.
Механізм виникнення синдрому відміни до кінця не виявлений. Вважають, що тканини «звикають» до високого рівня глюкокортикоїдних препаратів, на відміну яких вони реагують спалахом запальних процесів (вогнищева пневмонія, активація вогнищ хронічної інфекції тощо). Індукована глюкокортикоїдами недостатність кори надниркових залоз при цьому може відігравати певну роль, але далеко не головну.
З метою профілактики синдрому відміни рекомендується знижувати дози препаратів поступово. Лікування синдрому полягає у відновленні введення глюкокортикоїдних препаратів з подальшою більш повільною їх відміною. Вибір швидкості зниження дози багато в чому залежить від конкретної клінічної ситуації.
При тривалому застосуванні глюкокортикостероїдів (преднізолону, тріамцинолону та ін.) для лікування БА, захворювань сполучної тканини у хворих відмічають атрофію надниркової залози унаслідок пригнічення секреції кортикотропіну. Останній є фізіологічним стимулятором кори надниркової залози. Під час лікування глюкокортикоїдами для запобігання атрофії надниркової залози уводять кортикотропін або застосовують глюкокортикоїди з мінімальною резорбтивною активністю.
129. Гіперфункція кори надниркових залоз. Гіперфункція клубочкової зони кори – первинний та вторинний гіперальдостеронізм: етіологія, механізми та прояви.
Гіпе ральдостеронізм — це пат. стан, що виникає в результаті гіперфункції клубочкової зони кори надниркових залоз, яка продукує мінералокортикоїди.
Розрізняють первинний і вторинний гіперальдостеронізм.
Первинний гіперальдостеронізм (синдром Конна) виникає в результаті аденоми клубочкової зони, що утворює великі кількості альдостерону.
Основні прояви цього захв-ня:
1) АГ. Пов'язана зі збільшенням вмісту натрію у крові і стінках кровоносних судин, унаслідок чого підвищується чутливість їхніх гладких м'язів до дії пресорних факторів, зокрема катехоламінів;
2) гіпокаліємія (результат посиленої секреції іонів К+у канальцях нирок). Вона веде до порушень діяльності збудливих органів і тканин (порушення роботи серця, міастенія, парези);
3) негазовий алкалоз. Пов'язаний з посиленням ацидогенезу в дистальних звивистих канальцях нефронів;
4) поліурія. Виникає як наслідок втрати чутливості епітелію ниркових канальців до дії вазопресину (антидіуретичного гормону). Цим, зокрема, пояснюють той факт, що при первинному гіперальдостеронізмі ОЦК не зростає і набряки не розвиваються.
Вторинний гіперальдостеронізм є наслідком активації ренін-ангіотензинної системи, у процесі якої утворюються ангіотензини ІІ і III, що діють на кору надниркових залоз. Цей стан виявляється:
а) АГ;
б) набряками (гіперволемічними);
в) гіпокаліємією;
г) негазовим алкалозом.
130. Етіопатогенез спадкових порушень синтезу та периферичної дії альдостерону в дитячому віці.
Механізми регуляції синтезу і секреції надниркових андрогенів (ДЕА та андростендіона) щенедостатньо вивчені. Посилена секреція андрогенів наднирковими залозами розпочинається приблизно у віці 5-20 років (адренархе). Початок адренархе співпадає з диференціацією сітчастої зони у кірковійречовині надниркових залоз.
Вікові особливості клінічного перебігу надниркової недостатності.
Особливості клін. картини у ранньому віці.
Незрілість нирок та низька концентрація натрію вгрудному молоці і сумішах обумовлюють більшу потребу в альдостероні. Вираженими стаютьгіпонатріємія, гіпоглікемія, гіперкаліємія. Кетоз є не завжди, оскільки в цьому віці кетонові тілаутворюються у невеликій кількості. Гіперпігментація зазвичай відсутня, оскільки на її появу потрібнодекілька тижнів-місяців. Важко діагностувати артеріальну гіпотензію.
Хвороба розвивається стрімко. Важкі електролітні порушення з’являються вже за кілька діб післяпояви перших клін. ознак (зниження активності, відмова від грудей, блювота).
Особливості клінічної картини у старших дітей.
М’язова слабкість, втома, зниження апетиту,блювота, втрата маси тіла, ортостатична гіпотензія. Гіперпігментація є часто, але не завжди. Гіпоглікеміяз кетозом, гіпонатріємія. Гіперкаліємія розвивається пізніше. Клініка схожа до гастроентериту.
Хвороба розвивається поступово.
131. Гіперфункція пучкової зони кори наднирників. Хвороба та синдром Іценко-Кушинга. Прояви, пов’язані з надлишком глюкокортикоїдів. Патогенетичне обґрунтування діадіагностики первинного та вторинного гіперкортицизму та напрямків корекції.
Існує 2 клін. форми гіперфункції пучкової зони кори надниркових залоз.
1. Хвороба Іценка-Кушинга - базофільна аденома передньої частки гіпофіза.
2.Синдром Іценка-Кушинга:
а) пухлинний - аденома пучкової зони кори надниркових залоз;
б) ектопічна продукція АКТГ деякими злоякісними пухлинами (наприклад, рак легень);
в) ятрогенний — введення глюкокортикоїдів в організм з лікувальною метою.
Гіперфункція пучкової зони кори надниркових залоз виявляє себе утворенням і секрецією великих кількостей глюкокортикоїдів, високодозові ефекти яких і визначають клін. картину.
Для цього стану характерні:
1) АГ;
2) гіперглікемія — метастероїдшй ЦД;
3) ожиріння;
4) інфекційні захворювання з мінімальними ознаками запалення або без них;
5) шлункова гіперсекреція і утворення виразок у шлунку і 12-п.к.;
6) остеопороз;
7) м'язова слабкість;
8) уповільнене загоєння ран.
132. Патологія мозкової речовини наднирників. Феохромоцитома: причини і прояви.
Клітини мозкової речовини наднирникових залоз виробляють кетахоламіни – адреналін і норадреналін. Вони підвищують АТ, посилюють роботу серця, розширюють просвіти бронхів, збільшують рівень цукру в крові. В невеликій кількості виробляються пептиди і дофамін.
Патологія: Синдром Вотергауза-Фредеріксена, аденома наднирника, Аддісонова хвороба, потрійний-А синдром, синдром Іценка-Кушинга, синдром Конна, первинний гіперкортицизм, синдром Ашара-Тьєра, глюкостерома, первинний та вторинний гіперальдостеронізм, кортикоестрома, аденогенітальний синдром, феохромоцитома.
Причини феохромоцитоми: причини невідомі, зустрічається спорадично , але в 10% спостережень не виключається генетична природа захворювання.
Прояви: Хвороба проявляєтьсяхарактерниминападами з головнимболем, нудотою, тахікардією, підвищеним АТ, болем у підвилочковійділянці, з блювотою. Можуть бути посмикуванням'язіввсьоготіла, інодісудоми. Поза нападомхворіскаржаться на загальнуслабкість, втомлюваність, підвищенузбудливість, у окремихвипадках є постійнаартеріальнагіпертонія, а такожшлунково-кишковірозлади. Під час кризивідмічаютьпідвищеннярівняцукру у крові та лейкоцитоз. У сечі, особливо після нападу, підвищуєтьсявміст адреналіну, норадреналіну, ванілінмигдальноїкислоти.
133. Гіпотиреоз: класифікація, причини і механізми розвитку, патогенез основних порушень в організмі.
Виділяютьформи:
· Вроджений (первинний)
· Набутий (вторинний)
Останній, у свою чергу, поділяють на: первинний, вторинний, третинний і периферійний:
· первиннийгіпотиреоїдизмспостерігають при ураженнібезпосередньощитоподібноїзалози,
· вторинний є наслідкомпошкодження гіпофізу,
· третинний, пов'язанийзізмінами на рівнігіпоталамічнихцентрів,
· периферійний є наслідкомблокадициркулівнихтиреоїднихгормонівчирезистентності тканин до них.
Причинами розвиткугіпотиреоїдизму є:
· аплазія або гіпоплазія щитоподібноїзалози;
· генетичнодетермінованепорушеннябіосинтезутиреоїднихгормонів.
· оперативнихвтручаннях (тиреоїдектомія);
· рентгентерапії шиї (щитоподібної, паращитоподібнихзалоз, гортані), лікуваннірадіоактивнимйодом;
· прийманнілікарськихпрепаратів (зокрема, похіднихімідазола, препаратівлітія, йодидів, бета-блокаторів, інтерферону, інтерлейкіну-2);
· тиреоїдитах,
· деякихінфекційнихураженняхтаінфільтративнихзахворюванняхщитоподібноїзалози (туберкульоз, актиномікоз, рідше амілоїдоз, саркоїдоз, склеродермія, цистиноз, гемохроматоз);
· травмах щитоподібноїзалози, крововиливах, новоутвореннях;
· йододефіцитнихстанах
· черепно-мозковітравми з ураженням гіпофізу чи гіпоталамусу відповідно;
· пухлинита метастази;
· судинніпроцеси (крововиливи, тромбоемболії, атеросклероз);
· запальніпроцеси (аутоімунні та мікробні);
· хімічні інтоксикації, в тому числі й медикаментозні (резерпін, парлодел, апоморфінмаютьпереважнийвплив на гіпофіз).
Патогенез. При гіпотирозіпригнічуютьсявсівидиобмінів та утилізаціякисню тканинами, гальмуютьсяокислювальніреакції і знижуєтьсяактивністьрізнихферментних систем, газообмін і основнийобмін, порушуєтьсятерморегуляція. Порушеннябілковогообміну→уповільненнярозпаду і синтезу білка →порушенняобмінуглікозаміногліканів →накопичення в тканинах глікопротеїду муцина, гіалуронової та хондроїтинсірчаної кислот → при надлишкузмінюєтьсяколоїдну структуру сполучноїтканини → збільшенняїїгідрофільність і зв'язуваннянатрою, що в умовахутрудненоголімфовідтокуформуємікседему. На механізмзатримки в тканинах води і натріюможетакожвпливатинадлишоквазопресину, продукціяякогогальмуєтьсятироїдними гормонами. Порушенняліпідногообмінувиявляєтьсязниженням синтезу і розпадуліпідів. Підвищуєтьсявміст холестерину, тригліцеридів, бета-ліпопротеїдів. Порушуєтьсявуглеводнийобмін - сповільнюєтьсявсмоктуванняглюкозив кишечнику і їїутилізація в організмі. Дефіциттироїднихгормонівгальмуєрозвиток тканин мозку і пригнічуєвищунервовудіяльність. Розвиваєтьсягіпотироїднаенцефалопатія, при якійзнижуєтьсяінтелект і психічнаактивність, ослаблюєтьсяумовна і безумовна рефлекторна діяльність. Знижуєтьсяактивністьіншихзалозвнутрішньоїсекреції (зокрема, кора наднирників в умовахгіпотермії). Порушується і периферичнийметаболізмгормонівендокриннихзалоз (кортикостероїдів і статевихгормонів).
134. Гіпертиреоз: класифікація, причини і механізми розвитку, патогенез основних порушень в організмі.
При гіпертіроідізме рівень сироваткового T3 зазвичай вище, ніж рівень T4, що пов'язано, мабуть, з посиленою секрецією T3, а також конверсією T4 в T3 в периферичних тканинах. У деяких хворих підвищений рівень тільки T3 (T3-токсикоз).
T3-токсикоз зустрічається при гіпертиреозі, обумовленому будь-якими причинами - при хворобі Грейвса, многоузловом зобі і автономно функціонуючому солітарнийвузлі щитовидної залози. При відсутності лікування T3-токсикозу у хворого, як правило, є типові лабораторні ознаки гіпертиреозу (т. Е. Підвищення рівня T4 і поглинання 123I щитовидної залозою). Різні форми тиреоїдиту часто протікають дві фази: перша гіпертиреоїдних фаза змінюється гіпотиреоїдний.
Існує три форми захворювання:
Субклінічна. Явні симптоми відсутні, рівень Т4 в нормі, рівень трийодтироніну знижений;
Манифестная. Виявляються характерні ознаки гіпертиреозу. Рівень Т4 в нормі, рівень трийодтироніну низький;
Ускладнена. Симптоматика включає серцеву недостатність, аритмію, психоз та інші тяжкі стани.
За рівнем виникнення гіпертиреоз буває:
первинний - патологія щитовидної залози;
вторинний - уражається гіпофіз;
третинний - процеси розвиваються в гіпоталамусі.
Причини: До дисфункції щитовидної залози призводить ряд ендокринних захворювань, таких як:
Хвороба Грейвса (Базедова, Базедова-Грейвса). Синдром носить аутоімунний характер. В організмі виробляються антитіла, які стимулюють щитовидну залозу на вироблення надмірної кількості гормону Т4;
Вузловий зоб, токсична аденома. Перераховані патології супроводжуються формуванням доброякісних вузлів в тканинах залози. Освіти починають виробляти гормони і викликають гіпертиреоз. Лікарі поки не можуть сказати точно, чому одні аденоми синтезують Т4, а інші - ні;
Тиреоїдит. Запальний процес руйнує клітини щитовидки. Гормони потрапляють в кров і викликають гіпертиреоз. Можливий аутоімунний характер тиреоїдитів. В організмі виробляються антитіла проти рецепторів ТТГ. Клітини викликають активне зростання і запалення щитовидної залози.
135. Етіопатогенез вродженого гіпертиреозу, клінічні прояви.
Етіологія:
• зоб дифузний токсичний (Базедова хвороба) - найбільш часта причина гіпертиреозу
• зоб вузловий токсичний (хвороба Пламмер)
• підгострийтиреоїдит (тиреоїдит де Кервена)
• пухлини гіпофіза з надмірною секрецією ТТГ
• тератоми яєчників, що виробляють тиреоїдні гормони (Струма яєчника)
• гіперпродукція гормонів щитовидною залозою після надмірного введення в організм йоду (синдром йод-Базедов).
Патогенез:
• тиреоїднігормони збільшують споживання кисню тканинами, підвищуючи утворення тепла і енергетичний обмін
• підвищується чутливість тканин до катехоламінів і симпатичної стимуляції
• збільшується перетворення андрогенів в естрогени в тканинах і зростає вміст циркулюючого глобуліну, що зв'язує статеві гормони, що підвищує співвідношення естрогенів до андрогенів. Ці гормональні зміни можуть викликати гінекомастію у чоловіків
• швидке руйнування кортизолу під впливом тиреоїдних гормонів обумовлює клінічну картину гипокортицизма (оборотна надниркових залоз).
Клінічні прояви:
зміни метаболізму; спостерігається зниження ваги, незважаючи на хороший апетит і достатній прийом їжі; пітливість і непереносимість тепла; оборотна гіперглікемія; збільшення щитовидної залози; ЧСС збільшується; виникає стійка синусова тахікардія з частотою 120 в хв і бiльше (не зникає під час сну і погано піддається лікуванню серцевимиглікозидами) - хворий відчуває серцебиття в області шиї, голови та живота; симптоми хронічної серцевої недостатності; діарея; напади болю в животі; занепокоєння, дратівливість, патологічна відволікання, легкі порушення пам'яті, тремор; порушення менструального циклу; зниження потенції, можлива гінекомастія, м'язова слабкість і стомлюваність.
136. Зоб: види зобу, їх етіологія і патогенез; характеристика порушень функціонального стану залози, напрямки лікування та профілактики.
Етіпатогенетична
• Ендемічний зоб — спостерігають в ендемічних географічних районах по зобу.
• Спорадичний зоб — спостерігають в неендемічних районах по зобу.
Морфологічна
· Змішаний (дифузно-вузловий) зоб
За локалізацією
• Звичайнорозташований.
• Частковозагрудинний.
• Кільцевий.
• Дистопований зоб ізембріональних закладок (зоб кореняязика, пірамідальноїчасткищитоподібноїзалози).
Ендемічний зоб (нетоксичнийбагатовузловий зоб) виникаєвнаслідокдефіциту йоду в їжі. Нетоксичний зоб входить до ознак синдрому Ашера.
Дифузнийтоксичний зоб — автоімуннеураженнящитоподібноїзалози.
Гіпотиреоз
· Вродженіпорушення синтезу гормонівщитоподібноїзалози (див. кретинізм).
· Недостатністьвживанняйоду
· Антитиреоїднатерапія (наприклад, препаратом мерказоліл)
Гіпертиреоз
• Дифузнийтоксичний зоб (хвороба Грейвса)
• Тиреоїдити
Профілактика ендемічного зобу
1. Масова - застосування в осередках екзогенної йодної недостатності йодованої солі, що містить 25 г йодистого калію на 1 тонну.
2. Групова (дитячі, шкільні колективи, військові частини) за допомогою антіструміна (таблетка містить 1 мг йодистого калію), 1 таблетка на тиждень.
3. Індивідуальна - серед людей, які прибули в зону з йодною недостатністю застосовується антиструмин 1-2 таблетки на тиждень.
Поряд із специфічною профілактикою надзвичайно важливе значення має усунення поглиблюють факторів, які включають цілий ряд соціальних та санітарно-гігієнічних заходів (поліпшення житлових умов, повноцінне збалансоване харчування, профілактика гіповітамінозу, санітарна культура населення, боротьба з кишковими інфекціями і т.д.).
лікування зобу
1. Консервативне, як правило застосовується при дифузному зобі, або при змішаному (вузловому) при наявності абсолютних протипоказань до операції:
збалансоване харчування;
поліпшення санітарно-гігієнічних умов;
санація вогнищ хронічної інфекції;
нормалізація функції кишечника і печінки;
нормалізація обміну йоду в організмі: антиструмин 1 таблетка на добу (1 мг йодистого калію);
при необхідності призначення тиреоїдних гормонів 25 ... 100 мкг на добу; тиреокомб 0,5-1 табл. в день; тіреотом по 0,5-1 табл. в день, тиреоидин, трийодтиронін
2. Оперативне лікування зоба.
Показання до операції:
вузловий і змішаний зоб при всіх ступенях збільшення ЩЗ;
дифузний зоб III ст., що не піддається консервативної терапії.
дифузний зоб IV і V ст.
Характер оперативних втручань:
Резекція частки, можлива двостороння
геміструмектомія
137. Дифузно-токсичний зоб: причини, механізми, прояви та напрямки патогенетично обґрунтованого лікування
Дифузний токсичний зоб- гіпертиреоїдний (хвороба Гревса, базедова хвороба).
Характеризується ознаками гіперфункції щитоподібної залози, витрішкуватістю, підвищенням основного обміну, посиленням теплопродукції, тахікардією, тремтінням пальців рук, підвищенням психічної збудливості. У патогенезі базедової хвороби основного значення відіграє порушення імунологічних процесів і збільшення чутливості адренорецепторів до катехоламінів.
Негативний азотистий баланс при тиреотоксикозі свідчить про переважання катаболізму білків. Унаслідок посиленого розпаду глікогену в печінці та м’язовій тканині відзначається гіперглікемія.Надлишок тиреоїдних гормонів гальмує перехід вуглеводів у жири, прискорює розпад холестерину та його утилізацію в тканинах, інтенсифікує окиснення жирів у печінці, а також підвищує чутливість жирової тканини до ліполітичної дії адреналіну. Наслідком перелічених змін є посилена мобілізація жиру з депо, що пояснює схуднення хворих на тиреотоксикоз, гіпохолестеринемію та гіперкетонемії. Тиреоїдні гормони порушують метаболізм серцевого м’язу. Виявляються дистрофічні зміни в міокарді, порушення передсердно-шлуночкової провідності, перевантаження лівого шлуночка. Порушується енергетичне і пластичне забезпечення серцевої діяльності. «Тиреотоксичне» серце неадекватно реагує на холінергічні й адренергичні впливи.
Лікування при дифузному токсичному зобі спрямоване на зниження активності щитоподібної залози шляхом пригнічення синтезу тиреоїдних гормонів.
138. Порушення функції паращитоподібних залоз: причини, механізми, прояви.
При порушеннях функції паращитоподібних залоз виділяють гііпопаратиреоз та гіперпаратиреоз.
Причини гіпопаратиреозу -гіпофункції прищитоподібних залоз:
1) випадкове ушкодження або видалення прищитоподібних залоз при операціях на
щитоподібній залозі;
2) ушкодження прищитоподібних залоз при лікуванні радіоактивним йодом
хвороб щитоподібної залози;
3) аутоімунні ушкодження прищитоподібних залоз;
4) уроджене недорозвинення прищитоподібних залоз;
5) нечутливість клітин-мішеней до дії паратирину — псевдогіпопаратиреоз.
Основним проявом гіпопаратиреозу є гіпокальціємія. Вона обумовлює розвиток
паратиреопривної тетанії, що виявляється різким підвищенням нервово-м'язової
збудливості, множинними фібрилярними скороченнями м'язів усього тіла. Потім
виникають напади клонічних судом, що переходять у тонічні. Судомні скорочення
можуть поширюватися і на внутрішні органи (пілороспазм, ларингоспазм). Під час
одного з таких нападів настає смерть.
При хронічному гіпопаратиреозі у тварин розвивається клінічна картина парати-
реопривної кахексії. Вона характеризується схудненням, анорексією, підвищеною
нервово-м'язовою збудливістю, диспепсією й різноманітними трофічними
порушеннями.
Причини гіперпаратиреозу — гіперфункції прищитоподібних залоз:
1) пухлина — аденома прищитоподібної залози;
2) гіперфункція прищитоподібних залоз, обумовлена зменшенням чутливості їхніх ендокринних клітин до іонів кальцію, — порушення регуляції за принципом
негативного зворотного зв'язку.
Гіперпаратиреоз виявляється двома групами пов'язаних між собою змін.
І. Порушення кісткової тканини- генералізована фіброзна остеодистрофія.
Обумовлена підвищенням активності остеокластів і пригніченням функції
остеобластів. Виявляється болем у кістках і суглобах, розм'якшенням кісток,
різкою деформацією скелета. Розвивається демінералізація кісткової тканини
(остеомаляція), що обумовлює підвищення вмісту іонів кальцію в плазмі крові —
гіпер-кальціємію.
II. Гіперкальціємія. З нею пов'язані:
а) кальцифікація м'яких тканин (нирок, судин, легень). У важких випадках
розвивається ниркова недостатність;
б) утворення кальцієвих каменів у нирках;
в) порушення збудливості нервової системи і м'язів - м'язова слабкість, депресія,
порушення пам'яті;
г) артеріальна гіпертензія;
ґ) посилення шлункової секреції.
139. Порушення функції статевих залоз: первинні та вторинні стани гіпер- і гіпогонадизму.
Чоловічий гіпогонадизм — це гормональна недостатність чоловічих статевих за-
лоз. Його причинами можуть бути:
ї. Центральні (дисрегуляторні) порушення-зменшення утворення гонадолі-берину в
гіпоталамусі, лютеїнізуючого гормону в аденогіпофізі. Це може бути пов'язано з
ураженням зазначених структур, а також з гіперфункцією епіфіза, у разі якої
збільшення утворення мелатоніну супроводжується пригніченням синтезу
гонадотропних гормонів.
II. Власне залозисті порушення:
а) кастрація — видалення сім'яників;
б) фіброз яєчок після деяких вірусних інфекційних захворювань, ускладнених
орхітом (наприклад, після епідемічного паротиту);
в) порушення розвитку яєчок.
Після втрати яєчок розвивається комплекс порушень за назвою євнухізм.
Гіпофункцію яєчок при їх збереженні позначають терміном "євнухоїдизм".
III. Периферичні порушення:
а) зменшення чутливості клїтин-мтшенеи до дії андрогенів;
б) збільшене зв'язування тестостерону з білками плазми крові;
в) посилене руйнування андрогенів у печінці.
Прояви чоловічого гіпогонадизму залежать від того, у який період онтогенезу він
розвивається.
Виділяють такі групи причин жіночого гіпогонадизму.
I. Центральні (дисрегуляторнї) порушення. Можуть бути обумовлені
психогенними факторами, ураженнями гіпоталамуса (дефіцит гонадоліберину),
гіпофункцією аденогіпофіза (дефіцит ФСГ і ЛГ), гіперфункцією епіфіза
(зменшення утворення гонадотропних гормонів при збільшенні синтезу
мелатоніну).
II. Власне залозисті порушення:
а) спадково обумовлена аплазія яєчників;
б) дегенерація яєчників (запальна, кістозна);
в) аутоімунне ушкодження жіночих статевих залоз;
г) хірургічне видалення яєчників. III. Периферичні розлади:
а) зменшення чутливості клітин-мішеней до дії жіночих статевих гормонів;
б) підвищене зв'язування їх білками плазми крові;
в) посилене руйнування жіночих статевих гормонів у печінці.
Клінічні прояви гіпофункції статевих залоз залежать від часу настання гіпо-
гонадизму.
Якщо він розвивається до настання статевого дозрівання, то формується
комплекс порушень під назвою "оваріальний євнухоїдизм": слабкий розвиток
вторинних статевих ознак, первинна аменорея, високий зріст.
Розвиток гіпогонадизму в дітородному віці супроводжується порушеннями
циклічних процесів в організмі жінки (розлади менструального циклу),
безплідністю, передчасним клімаксом.
Після менопаузи розвиваються клімактеричні зміни - нестабільність судинного
тонусу, остеопороз та ін.
Чоловічий гіпергонадизм може розвиватися в дитячому віці до періоду статевого
дозрівання і у дорослих.У дітей гіпергонадизм найчастіше пов'язаний з пухлинами і запальними процесами
в гіпоталамусі, а також гіпофункцією епіфіза. Усі ці причини викликають
збільшення продукції гонадотропних гормонів і, як наслідок, андрогенів. Клінічно
такий гіпергонадизм виявляється передчасним статевим дозріванням.
Причиною гіперфункції статевих залоз у дорослих є добро- і злоякісні пухлини, що
ростуть з інтерстиціальних клітин. При цьому виражених клінічних ознак,
пов'язаних з гіперандрогенемією, немає. У випадку злоякісних новоутворень
характерні відсутність кахексії, збереження м'язової маси і сили м'язів
В основі жіночого гіпергонадизму, при якому збільшується секреція жіночих
статевих гормонів, можуть лежати центральні (дисрегуляторні) порушення і власне
залозисті причини.
Центральні порушення, обумовлені збільшенням секреції гонадоліберину і
гонадотропних гормонів, можуть виявлятися розвитком синдрому передчасного
статевого дозрівання і синдрому уявної вагітності.
Для першого характерні рання поява вторинних статевих ознак, ранній початок
менструацій і ймовірність завагітніти (у віці 7—8 років). Другий синдром
виявляється аменореєю й усіма зовнішніми ознаками вагітності. При цьому має
місце підвищена продукція пролактину клітинами аденогіпофіза.
До власне залозистих причин жіночого гіпергонадизму відносять кістозні процеси і
пухлини яєчників, що продукують гормони. При цьому розвивається гіпертрофія
ендометрію, порушується менструальний цикл, поновлюються маткові кровотечі в
менопаузі.
140. Класифікація та патогенез адреногенітального синдрому у дитячому віці.
Група спадкових захворювань, що виникають внаслідок дефіциту ферментів, які беруть участь в утворенні гормонів надниркових залоз.
Класифікація:
- солевтратна форма;
- гіпертонічна форма;
- проста вірильна форма:
• вроджена, класична;
• періоду пубертату (некласична форма).
Патогенез
Обумовлюється гіперпродукцією андрогенних і естрогенних гормонів, а також їхніх біохімічних метаболітів, які мають різну гормональну активність. Кількість виділених стероїдних гормонів безпосередньо залежить від виду патологоанатомічних змін у корі надниркових залоз — гіперплазії або пухлини — і від секреторної активності. Найчастіше він спричинюється природженою дифузною гіперплазією сітчастої зони кори надниркових залоз, рідше — доброякісною або злоякісною пухлиною кори. Синдром може передаватися спадково як рецесивна ознака, при якій носії мають порушений стероїдосинтез. Для природженої форми цього захворювання характерно те, що ознаки його з'являються одразу після народження. Зазвичай переважає секреція андрогенних гормонів, що обумовлює розвиток вірилізму. Внаслідок цього у жінок андрогенна активність розвивається в напрямку гетеросексуалізму, а у чоловіків — ізосексуалізму. Набагато рідше буває переважання підвищеної продукції естрогенів, які обумовлюють розвиток гомосексуальних явищ у жінок, і гетеросексуальних явищ у чоловіків, що виявляються фемінізацією. Надниркові залози починають функціювати ще в період внутрішньоутробного розвитку плода, через це при адреногенітальному синдромі вироблення надлишку андрогенних і естрогенних гормонів впливає на вже сформований у статевому відношенні плід. Зміни статевої системи, первинних і вторинних статевих ознак тим виразніші, чим раніше під час розвитку плода розвинулася підвищена функція кори надниркових залоз.
141. Етіопатогенез та прояви синдрому тестикулярної фемінізації у дітей та підлітків.
форма чоловічого псевдогермафродитизму, яка найчастіше спостерігається серед інших аномалій розвитку, зумовлена дефектом андрогенних рецепторів і характеризується каріотипом 46ХY, наявністю тестикул, жіночими або частково маскулінізованими зовнішніми статевими органами. СТФ розділяють на дві клінічні підгрупи, зважаючи на генітальний фенотип: 1) повна форма синдрому; 2) неповна форма синдрому. СТФ є зчепленою з Х хромосомою ознакою.
Патогенез
СТФ зумовлений дефектом гена рецептора до андрогенів, який розташований на короткому плечі Х хромосоми. У процесі ембріогенезу в цих осіб під впливом Y хромосоми гонади диференціюються як яєчка, які секретують тестостерон і речовину, що інгібує протоки Міллера. Проте через дефект гена андрогенних рецепторів відсутня чутливість до тестостерону і дигідротестостерону, відповідальних за формування чоловічого фенотипу. Таким чином, закономірно формується жіночий фенотип за відсутності похідних проток Міллера.
Хворі відрізняються жіночою статурою з добре розвиненими грудними залозами, незначним лобковим і паховим оволосінням, відсутністю внутрішніх статевих органів і «вагінальним мішком», який закінчується сліпо. Глибина
піхви може варіювати від нормальної або вкороченої, аж до наявності вагінальної заглибини глибиною 1–2 см. Неповний варіант схожий на повну форму СТФ за винятком ознак маскулінізації (вірилізації) зовнішніх статевих органів і наявністю статевого оволосіння.
142.Стрес. Визначення поняття, причини і механізми розвитку, стадії. Поняття про “хвороби адаптації".
Терміном "стрес" позначають неспецифічну реакцію організму, що виникає під впливом будь-яких сильних впливів і супроводжується перебудовою захисних систем організму. Стрес проявляється у вигляді загального адаптаційного синдрому, який складається з трьох послідовних стадій: реакції тривоги, стадії резистентності і стадії виснаження.
• Реакція тривоги означає негайну мобілізацію захисних сил організму. Вона складається з фази шоку і протівошока. У фазі шоку спостерігаються гіпотонія м'язів і артеріальна гіпотензія, гіпотермія, гіпоглікемія, згущення крові, еозинопенія, підвищення проникності капілярних судин. Інволюція лімфоїдної тканини, негативний баланс азоту, виразкові ураження шлунка свідчать про переважання процесів катаболізму. Фаза протівошока характеризується змінами в зворотному напрямку, що ведуть до розвитку наступної стадії - стадії резистентності. Основне патогенетичне ланка фази протівошока - це стійке посилення секреції кортикотропіну і кортикостероїдів.
• У стадії резистентності гіпертрофується кіркова речовина надниркових залоз і секретується велика кількість гормонів, активізуються анаболічні процеси, посилюється гліконеогенез.
• При тривалій дії пошкоджуючого агента адаптація порушується. Виснаження функціональних резервів і атрофія коркового речовини надниркових залоз, зниження артеріального тиску, розпад білкових речовин характеризують перехід стадії резистентності в стадію виснаження. Результат стресу залежить від співвідношення сили і тривалості дії стресора і потенційних можливостей захисних сил організму. Біологічне значення адаптаційного синдрому полягає не тільки в тому, що в другій, найбільш тривалій його стадії підвищується резистентність організму по відношенню до фактору, який викликав стан стресу, але і в тому, що при не дуже сильному і тривалому стресі може створюватися або підвищуватися неспецифічна резистентність організму до різних інших факторів.Недостатність адаптації або її відхилення в протилежну сторону є причиною розвитку хвороб адаптації. До хвороб адаптації відносяться ревматизм, бронхіальна астма, деякі хвороби нирок, серця і судин, ряд шкірних та інших захворювань.
143.Особливості розвитку типових процесів в нервовій системі патологічних
Порушення діяльності нервової системи можливі в результаті розвитку типових патологічних процесів - запалення, пухлини, місцевих порушень кровообігу.
Пухлина подразнює той чи інший нервовий центр, викликаючи його надмірне збудження. У міру ж росту пухлини розвивається атрофія нервових клітин і волокон, що призводить до вимикання їх функцій. Крім того, збільшення маси пухлини супроводжується підвищенням внутрішньочерепного тиску, - зменшенням кровонаповнення головного мозку і його ішемією.
Запалення досить часто є причиною порушення функцій нервової системи. Запалення периферичних нервових провідників супроводжується порушеннями чутливості, руху або діяльності внутрішніх органів. Запалення, що виникло в центральній нервовій системі, найчастіше локалізується в мозкових оболонках і призводить до порушення продукції і відтоку спинномозкової рідини, підвищення внутрішньочерепного тиску, порушення мозкового кровообігу. Запалення може захоплювати і речовину мозку. У патогенезі енцефаліту істотну роль грає аутоаллергических реакція, оскільки у організму відсутня імунологічна толерантність до власної нервової тканини. Що стосується патогенезу порушень діяльності нервової системи, то треба зазначити, що досить добре вивчені лише механізми порушень функцій нейронів. Такими універсальними механізмами є втрата нервовою клітиною здатності підтримувати певну величину мембранного потенціалу, генерувати потенціали дії і проводити їх по відростках, передавати збудження з однієї нервової клітини на іншу.
Інтегративні та аналітичні здібності нервової системи багато в чому визначаються множинними контактами нервових клітин один з одним. Зменшення кількості межнейрональних контактів в процесі розвитку ряду патологічних процесів, також є одним з істотних механізмів порушення функції нервової системи. Важливою ланкою в патогенезі багатьох розладів діяльності нервової системи може бути порушення виділення і розпаду медіаторів.
Говорячи про загальні закономірності порушень діяльності нервової системи, слід сказати, що в патогенезі функціональних порушень її центральних відділів може мати значення поява в нервових центрах групи нейронів, які працюють з тим чи іншим ступенем автономності та продукують надмірне збудження. У нормі активність нейронів або нервових центрів контролюється і обмежується відповідними механізмами гальмування. При пошкодженні цих механізмів і виникає генератор патологічно посиленого збудження, який в залежності від його локалізації може бути причиною чутливих, рухових і вегетативних розладів, а також порушень вищої нервової діяльності.
144.Порушення сенсорної функції нервової системи
Патологічні процеси і пов'язані з порушеннями чутливості можуть локалізуватися на будь-якій ділянці сенсорного шляху. При пошкодженні периферичних нервів у відповідній зоні порушуються всі види чутливості. Втрата чутливості називається анестезією, зниження - гіпестезією, підвищення - гиперестезией. Залежно від характеру втраченої чутливості розрізняють анестезію тактильну, больову, термічну, а також втрату пропріоцептивної, чутливості.
Існує дві доцентрові системи чутливості. Одна з них називається лемнисковій і містить нервові волокна великого діаметра, які проводять імпульси від проприорецепторов м'язів, сухожиль, суглобів і частково від шкірних тактильних рецепторів. Волокна цієї системи входять в спинний мозок і йдуть в складі задніх стовпів в довгастий мозок. Від ядер довгастого мозку починається медіальна петля, яка переходить на протилежну сторону і закінчується в заднебокових вентральних ядрах таламуса, нейрони яких передають отриману інформацію в соматосенсорную зону кори великого мозку.
Друга висхідна система - це спиноталамический шлях, який несе больову, температурну і частково тактильну чутливість. Волокна його йдуть вгору в складі передніх і бічних канатиків спинного мозку і закінчуються в клітинах ядер таламуса.
Вельми характерні зміни чутливості спостерігаються при перерезке правої або лівої половини спинного мозку: на стороні перерізання нижче її зникає глибока чутливість, в той час як температурна і больова зникають на протилежній стороні, оскільки провідні шляхи, які стосуються антеролатеральной системі, перехрещуються в спинному мозку. Тактильна чутливість частково порушена по обидва боки.
Порушення лемнискової системи можливо при пошкодженні периферичних нервів, а також при різних патологічних процесах в спинному мозку. Ізольоване ураження задніх канатиків спинного мозку зустрічається рідко, але поряд з іншими провідними шляхами вони можуть бути пошкоджені пухлиною або під час травми.
Порушення провідності в волокнах медіальної петлі викликає різні порушення чутливості, вираженість яких залежить від ступеня пошкодження системи. При цьому може втрачатися здатність визначати швидкість і напрямок руху кінцівок. Значно порушується почуття роздільного сприйняття дотиків одночасно в двох місцях, а також здатність відчувати вібрацію і оцінювати тяжкість вантажу, що піднімається. Випробуваний не може на дотик визначити форму предметів і ідентифікувати букви і числа, якщо написати їх на шкірі: він відчуває тільки механічне дотик і не може точно судити про місце і силі тактильного відчуття. Відчуття болю і температурна чутливість при цьому зберігаються.
Пошкодження постцентральна звивини кори великого мозку. У людини ізольоване ураження постцентральна звивини буває дуже рідко. Наприклад, хірурги іноді видаляють частину цієї звивини для лікування епілепсії коркового походження. В цьому випадку втрачається відчуття положення кінцівок в просторі, здатність на дотик визначати форму предметів, їх розміри, масу, характер поверхні, втрачається дискримінаційна чутливість.
145.Біль. Принципи класифікації. Сучасні уявлення про причини і механізми розвитку болю: теорія інтенсивності, теорія специфічності. Патологічний біль. Реакції організму на біль. Природні антиноцицептивні механізми.
У поняття болю включається, по-перше, своєрідне відчуття і, по-друге, реакція на болюче відчуття, яка характеризується певною емоційним забарвленням, рефлекторними змінами функцій внутрішніх органів, руховими безумовними рефлексами і вольовими зусиллями, спрямованими на позбавлення від больового фактора. Спостереження показують, що при дії шкідливого чинника людина може відчувати два різновиди болю. "Перша" біль - чітко локалізована і швидко стихає. "Друга" біль - може тривати досить довго.
Значне місце в симптоматиці різних хвороб займає вісцеральний біль. Цей біль насилу піддається чіткій локалізації, носить розлитої характер, супроводжується тяжкими переживаннями, пригніченням, пригніченістю, зміною діяльності вегетативної нервової системи. Складним і поки ще не вирішеним є питання про те, які нервові освіти беруть участь в рецепції, проведенні та сприйнятті болю. З цього питання існує дві принципово різні точки зору. Згідно з однією з них, біль не є специфічним, особливим почуттям і не існує спеціальних нервових приладів, що сприймають тільки болюче подразнення. Будь-яке відчуття, засноване на подразненні тих чи інших рецепторів, може перейти в біль, якщо сила роздратування досить велика і перевершила відомий межа.
Відповідно до іншої точки зору, існують спеціальні больові рецептори, спеціальні аферентні шляхи, що передають болюче подразнення, і спеціальні структури в головному мозку, які переробляють больову інформацію.Дослідження показують, що рецептори шкіри і видимих слизових, що реагують на больові стимули, належать до двох типів чутливих волокон антеролатеральной системи - тонким мієлінових АД-волокнам і неміеліновим С-волокнам. Активність в тонких мієлінових АД-волокнах викликає у людини відчуття гострої колючого болю, тоді як збудження повільно проводять С-волокон викликає відчуття печіння.
Питання про механізми активації больових рецепторів поки ще остаточно не з'ясовано. Є припущення, що сама по собі сильна деформація вільних нервових закінчень, служить адекватним стимулом для рецепторів болю, впливає на проникність клітинної мембрани в них і призводить до виникнення потенціалу дії. Відповідно до іншої гіпотези, вільні нервові закінчення, які стосуються АТ- або С-волокнам, містять одну або кілька специфічних речовин, які виділяються під дією механічних, термічних та інших факторів, взаємодіють з рецепторами зовнішньої поверхні мембрани нервових закінчень і викликають їх збудження. Надалі ці речовини руйнуються відповідними ферментами, оточуючими нервові закінчення, і відчуття болю зникає. Як активаторів ноцицептивних рецепторів запропоновані гістамін, серотонін, брадикінін, соматостатин, субстанція Р, простагландини, іони К +.
Фантомний біль виникає у людей після ампутації кінцівок. Протягом тривалого часу хворий може відчувати ампутовану кінцівку і сильну, часом нестерпний біль у ній. При ампутації зазвичай перерізаються великі нервові стовбури з великою кількістю товстих нервових волокон, перериваються канали для надходження імпульсації з периферії. Нейрони спинного мозку стають менш керованими і можуть давати спалахи на найнесподіваніші стимули. Каузалгія - жорстока, болісна біль, що спостерігається при пошкодженні будь-якого великого соматичного нерва. Будь-яке, навіть саме незначне вплив на хвору кінцівку викликає різке посилення болю. Каузалгія виникає частіше в разі неповної перерізання нерва, коли ушкоджується велика частина товстих мієлінових волокон.
При розвитку в деяких внутрішніх органах патологічних процесів може виникати відбитий біль. Відображена біль пояснюється тим, що ушкодження внутрішніх органів викликає збудження, яке по аферентних волокнах вегетативних нервів досягає тих же нейронів задніх рогів спинного мозку, на яких закінчуються аферентні волокна від шкіри. Посилена аферентна пульсація від внутрішніх органів знижує поріг збудливості нейронів таким чином, що роздратування відповідної ділянки шкіри сприймається як біль.
Експериментальні та клінічні спостереження вказують на те, що у формуванні больового відчуття і реакції організму на біль беруть участь багато відділів центральної нервової системи. Через спинний мозок реалізуються моторні і симпатичні рефлекси, там же відбувається первинна обробка больових сигналів. Ретикулярна формація виконує підготовку і передачу больовий інформації до вищих соматичні і вегетативні відділи головного мозку, полегшення захисних сегментарних рефлексів спинного мозку і стовбура мозку, залучення в рефлекторний відповідь на больові стимули вегетативної нервової системи, дихального і гемодинамічного центрів. Зоровий бугор забезпечує аналіз якості больового відчуття. Больова інформація активує нейрогенні і нейрогормональні структури гіпоталамуса. Це супроводжується розвитком комплексу вегетативних, ендокринних і емоційних реакцій, спрямованих на перебудову всіх систем організму в умовах дії больових стимулів. Лімбічна система відіграє важливу роль у створенні емоційного забарвлення поведінки організму у відповідь на больову стимуляцію. Мозочок, пірамідна і екстрапірамідна системи здійснюють програмування рухових компонентів поведінкових реакцій при виникненні больового відчуття. За участю кори реалізуються свідомі компоненти больового поведінки.
Антиноцицептивні системи мозку. У нервовій системі є не тільки больові центри, збудження яких веде до формування больового відчуття, але і структури, активізація яких здатна змінити больову реакцію у тварин аж до її повного зникнення. Є докази того, що таких систем в мозку чотири:
1) нейронна опіатна;
2) гормональна опіатна;
3) нейронна неопіатного;
4) гормональна неопіатного.
Значення болю для організму. Біль викликається різними факторами, єдиним загальною властивістю яких є здатність пошкоджувати тканини організму. Вона відноситься до категорії патологічних процесів і як будь-який патологічний процес суперечлива за своїм змістом. Біль має як захисно-пристосувальне, так і патологічне значення. Залежно від характеру болю, причини, часу і місця її виникнення можуть переважати або захисні, або власне патологічні елементи. Значення захисних властивостей болю справді величезне для життя людини і тварин: вони є сигналом небезпеки, інформують про розвиток патологічного процесу.
146.Порушення рухової функції нервової системи
Порушення γ-мотонейронів призводить до скорочення веретен, що супроводжується збільшенням в них частоти імпульсації, яка по аферентні волокнах досягає α-мотонейронів. Наслідком цього є порушення α-мотонейронів і підвищення тонусу відповідних м'язів.
Рухові розлади виникають як при пошкодженні зазначених відділів центральної нервової системи, так і при порушенні проведення імпульсів по рухових нервах і передачі імпульсів з нерва на м'яз.
Найбільш поширеною формою рухових порушень є параліч і парез - втрата або ослаблення рухів внаслідок порушення рухової функції нервової системи. Параліч м'язів однієї половини тіла називається гемиплегией, обох верхніх або нижніх кінцівок - параплегией, всіх кінцівок - тетраплегія. Залежно від патогенезу паралічу тонус уражених м'язів може бути або втрачений, або підвищений. Крім того, розрізняють параліч периферичний і центральний.
При ураженні рухових нервів в іннервіруємих м'язах розвивається параліч, зникають всі рефлекси, вони атонічная і з плином часу атрофуються. В експерименті такий тип рухових розладів зазвичай отримують шляхом перерізання передніх спинномозкових корінців або периферичного нерва.
Особливий випадок являє собою рефлекторний параліч, зумовлений тим, що при пошкодженні будь-якого чутливого нерва імпульси, які виходять від нього, можуть надавати гальмівну дію на мотонейрони відповідної м'язи.
147.Порушення трофічної функції нервової системи. Нейрогенні дистрофії. Етіологія, патогенез.
Нейродистрофічним називається комплексний процес, за якого внаслідок випадіння чи порушення різних нервових впливів як соматичної, так і вегетативної нервової системи виникають розлади її трофічної функції з наступним розвитком в організмі складних змін, а саме:
1) структурних розладів — дистрофічні зміни, некроз і виразкування, атрофія;
2) функціональних змін - підвищення чутливості денервованих структур до дії гуморальних факторів;
3) метаболічні розлади — зміна інтенсивності, патологічне пригнічення чи активація ферментних систем, поява біохімічних процесів ембріонального характеру.
В основі нейродистрофічного процесу – зміни синтезу, секреції чи дії нейромедіаторів, комедіаторів, трофогенів, а також утворення патотрофогенів, тощо; посилюється при розладах гемо- та лімфоциркуляції, енергетичного та пластичного обмінів. Трофічні розлади виникають у будь-якому органі, якщо порушується його іннервація.
Причини і механізми порушень електрофізіологічних процесів в нейронах. Порушення діяльності іонних каналів. Причини та механізми порушень нейрохімічних процесів. Порушення обміну нейротрансмітерів, нейромодуляторів, нейрогормонів. Патологічне збудження і патологічне гальмування нервових центрів. Неврози.
Пошкодження нейронів як одна з причин порушень інтегративних функцій ЦНС.
Гострі і хронічні розлади мозкового кровообігу. Інсульт. Набряк і набухання головного мозку, причини і механізми розвитку. Внутрішньочерепна гіпертензія. Роль ушкоджень нейроглії в розвитку патологічних процесів у ЦНС. Пошкодження гематоенцефалічного бар’єра та аутоімунні ураження головного мозку.
148.Етіопатогенез та принципи лікування перинатальної гіпоксично-ішемічної енцефалопатії
Перинатальна гіпоксично-ішемічна енцефалопатія (ГІЕ) — поширена патологія нервової системи як серед доношених, так і серед недоношених новонароджених. Лікування ГІЕ у новонароджених є складним і дискута- бельним. Перинатальна гіпоксично-ішемічна енцефалопатія (ГІЕ) зумовлена недостатнім надходженням кисню в тканини мозку, що пов’язане як зі зниженням вмісту кисню в артеріальній крові, так і зі зменшенням мозкового кровообігу [27]. Це призводить до каскаду патологічних процесів у ішемізованій тканині мозку: глутамат-кальцієвої ексайтотоксичності, запалення та оксидантного стресу [27]. Такі несприятливі події в мозку новонародженого впливають на подальший розвиток дитини і призводять до тривалих тяжких неврологічних дефектів, таких як розумова недостатність, мовні порушення, труднощі у навчанні, рухові розлади тощо [10].
Перинатальна ГІЕ зазвичай клінічно виявляється в перші дні життя новонародженого. Її вияви — складність ініціювання і підтримки дихання, нестабільність температури тіла, зміна м’язового тонусу, пригнічення фізіологічних рефлексів та зміна рівня свідомості [10]. У новонароджених ГІЕ спричиняє судоми різного характеру [36].
Серед перинатальних чинників ризику розвитку ГІЕ найбільше значення мають гіпоксія та ішемія головного мозку як наслідок системної гіпоксемії і зниження мозкового кровообігу, внаслідок чого виникають структурні ушкодження головного мозку. Зменшення мозкової перфузії спричиняє ураження білої речовини перивентрикулярної ділянки у недоношених дітей і кори головного мозку — у доношених [1]. Гіпоперфузія мозку призводить до лактат-ацидозу та розвитку енергетичного дефіциту. Виділяють декілька фаз енергетичної недостатності при ГІЕ [8]. лікування наслідків ГІЕ, мають передбачати застосування агентів, які впливають на вільнорадикальні процеси, переривають реакції глутамат-кальцієвого каскаду, блокують дію прозапальних цитокинів, гальмують прооксидантні ферменти, посилюють трофічне забезпечення та стабілізують процеси апоптозу.
149.Старіння. Сучасні теорії старіння, засоби продовження активного життя за О.О. Богомольцем.
Старіння – процес суперечливого розвитку живих клітин від моменту зародження життя до його закінчення. Припущення, що потенційно безсмертні мікроорганізми, позбавлені наукової цінності життя планети як ціле – безмежне,можливостям її розвитку немає меж, але життя в рамках індивідуального існування, обмеженого в часі, не володіє цими властивостями. Для етапу індивідуального розвитку життєві програми запрограмовані, включаючи старіння і закінчення життя-смерть.
Основні поняття геронтології – вік, старіння, старість, довголіття, безсмертя – відображають людські уявлення про життєві процеси, об’єктивні закони життя, а також сильне бажання жити довго.
Загальні закономірності й теорії старіння
Існує дві традиційні точки зору на причини розвитку старіння (закономірності):
Старіння – генетично запрограмований процес, результат закономірної реалізації програми, закладеної у генетичному апараті.
Старіння – результат руйнування організму, викликаний різними факторами, дія яких повторюється і накопичується на протязі всього життя.
теорії процесу старіння
Вихідною позицією представників так званої теорії зношування є: живі системи старіють під впливом інтенсивних життєвих процесів, а старіння прискорюється чи сповільнюється за законами фізики в залежності від динаміки процесів на рівні клітини, тканин, цілого організму. Вчені цієї теорії доводили, що всі індивіди в популяції мають приблизно однакову тривалість життя, але її межа визначається темпом зношування, при цьому тривалість життя залежить від середньої величини витраченої енергії на 1 кг маси індивіда.
Теорія витрачення "життєвої" енергії в клітинах. Представники цієї теорії старіння (М. Бергер) вважають, що кожний організм отримує у спадщину визначену кількість "життєвого фермента", який з часом витрачається, що наближує організм до смерті.
Математична модель старіння і старості дозволила досліджувати старість як закономірні, математичні вимірювані явища, що протікають з поступовим збільшенням захворювань і ймовірності смерті.
Інтоксикаційні теорії старіння. В кінці XIX ст. Ч. Бухард висунув положення "Кожен організм є лабораторією для токсинів". Вчені вважають, що старіння представляє собою процес само-інтоксикації в результаті збільшення рівня токсинів у клітині.
Теорію дисгармонії представляє концепція внутрішніх протиріч, згідно якої старіння є результатом порушення можливості оновлення клітини.
Існує концепція, яка пояснює старіння впливом біофізичних факторів на генетичний апарат клітин і накопичення радіоактивних речовин. Висока концентрація радіоактивних елементів порушує обмінні процеси і викликає процеси старіння в клітині.
Суть молекулярних і клітинних механізмів старіння полягає у:
1. Порушенні генетичного апарату клітин, програми біосинтезу білка (з віком накопичуються помилки в генетичній інформації, що призводить до появи "дефектних" білків).
2. Порушення клітинної біоенергетики.
3. Зменшення клітинної маси (відмирання певної частини клітин призводить до того, що на інші клітини випадає велике навантаження, що сприяє їх гіперфункції; викликає старіння).
4. Цитоморфологічні зміни (в клітинах, які не діляться накопичуються продукти їх життєдіяльності, що сприяє старінню).
5. Функціональні зміни:
зниження здатності нейронів відтворювати інформацію;
знижується функція секреторних клітин – синтезувати і виділяти речовини;
зниження рівня працездатності, та ін.
150.Екстремальні стани, визначення поняття, види екстремальних станів.
Екстремальні – це стани організму, які хар. надмірним напруженням чи виснаженням пристосувальних механізмів. Можуть розвиватись первинно під дією надзвичайних подразників (травм, інтоксикацій, різких коливань температури чи вмісту О2) або бути результатом несприятливого перебігу захворювання (недостатності кровообігу, дихання, нирок, печінки, анемії тощо). Найбільш частими є шок, колапс та кома. Етіологія. Екстремальні стани розвиваються під впливом екстремальних факторів, які відрізняються від інших патогенних агентів украй високим, гранично інтенсивним, часто руйнівним ефектом. Екстремальні фактори поділяються на: 1) екзогенні й 2) ендогенні. Екзогенні екстремальні фактори можуть мати: а) фізичну, б) хімічну чи в) біологічну природу. Фактори фізичної природи: механічні, електричні, термічні, барометричні, радіаційні, гравітаційні. Хімічні фактори: граничний дефіцит чи надлишок кисню, субстратів метаболізму, рідини; виражені інтоксикації ліками, промисловими отрутами, кислотами, лугами. Біологічні фактори: значний дефіцит чи надлишок екзогенних БАР; мікроби, паразити і гриби. Ендогенні екстремальні фактори: а) несприятливий, важкий плин хвороб і хворобливих станів; б) виражена недостатність функцій органів і фізіологічних систем; в) значна крововтрата; г) масивні крововиливи в органи; д) надлишок продуктів імунних чи алергійних реакцій; е) істотний дефіцит чи надлишок БАР або їх ефектів; є) психічні перенапруги і травми. Умови, що сприяють виникненню екстремальних станів. 1) Фактори, що потенціюють ефекти екстремальних агентів: а) тривале голодування; б) нервове чи психічне перенапруження; в) значна фізична втома; г) загальні гіпер- чи гіпоергичні стани, д) хронічні важкі хвороби; е) переохолодження і перегрівання організму. 2) Реактивність організму часто є вирішальною умовою при дії екстремального фактора. На відміну від нормергічного реагування гіпер- чи гіпоергічний стан організму істотно полегшує виникнення, усугубляє перебіг і завершення екстремального стану. Від екстремальних необхідно відрізняти термінальні стани, що є кінцевими етапами життя організму, прикордонними станами між життям і смертю. Найбільш важливими екстремальними станами і такими, що найчастіше зустрічаються, є шок, колапс, кома.
151. Шок. Визначення поняття, етіологія шокових станів. Види шоків.
Шок – це тяжкий патол.процес, із виснаженням життєво важливих ф-й орг. І веде його на грань між смертю та життям через критичне зменшення капілярного кровообігу в уражених органах. Види: А) за причинами: травматичний, геморагічний, опіковий, турнікетний (після джгута), ангідремічний, кардіогенний, панкреатичний, анафілактичний, септичний, інфекційно-токсичний. Б)за первинними механізмами вин.: 1)гіповолемічний (геморагічний, ангідремічний), 2)повяз.з насосною ф-ю серця (кардіогенний), 3)судинні форми (анафілактичний, панкреатичний) 4)больовий (травматичний, опіковий). Механізми: пат.ланка – гостра недостатність кровообігу (зниж.АТ, ХОК, пор.реології крові тощо), її механізми: 1)зменш.обєму циркулюючої крові (крововтрата, масивне ексудативне запалення при опіку, збільшення проникності судин при анафілаксії, зневоднення, перерозподіл крові внаслідок тромбозу чи емболії). 2)Зменшення ХОК (змен.скоротливості при інфаркті, тампонада серця при розриві чи ексуд.перикардиті, фібриляція шлуночків) 3)Змен.заг.пер. опору (зниження нейрогенного тонусу судин при больовому шоці, зменшення баз.тонусу під впливом БАР (анафілаксія, панкреатич.шок) чи токсинів (травма, турнікетний, інфекційно-токсичний.) 4)порушення реології крові (ДВЗ синдром при панкреатичному шоці, агрегація при сепсисі чи інф.токсичному шоці, гемоконцентрація при зневодненні). Наслідки: порушення кровообугу (мозкового, вінцевого, ниркового) це веде до розвитку коми та гострої недостатності ССС та нирок. Приєднуються гіпоксія, ацидоз та інтоксикація. Травматичний шок (стадія збудження та торпідна ст.).
152.Загальні елементи патогенезу шокових станів. Порушення нейро-ендокринної системи, порушення системної гемодинаміки та мікроциркуляції. Клітинні порушення при шоках. «Шокові» органи.
Незалежно від причин виникнення шок виявляє себе комплексом порушень гемодинаміки, для якого характерні зменшення артеріального тиску, хвилинного об'єму серця, венозного повернення до серця, об'єму циркулюючої крові, об'ємної швидкості органного кровообігу; порушення реологічних властивостей крові (агрегація формених елементів, підвищення в'язкості крові). Комплекс цих порушень може бути визначений як гостра недостатність кровообігу.
Відповідно до законів гемодинаміки первинне порушення одних її показників при будь-якому різновиді шоку веде вторинно до порушень усіх інших.
В основі розвитку розладів кровообігу за умов шоку можуть лежати такі механізми.
I. Зменшення об'єму циркулюючої крові:
1) крововтрата (геморагічний шок);
2) втрата плазми крові при великому ексудативному запаленні (опіковий шок);
3) вихід рідини з кровоносних судин у тканини при генералізованому підвищенні проникності судин (анафілактичний шок);
4) зневоднення (ангІдремічний шок);
5) перерозподіл крові в судинному руслі (тромбоз і емболія магістральних вен).
II. Зменшення хвилинного об'єму серця:
1) порушення скоротливої функції серця (інфаркт міокарда);
2) тампонада серця (розрив серця, ексудативний перикардит);
3) аритмії (фібриляція шлуночків).
III. Зменшення загального периферичного опору - генерал із оване розширення судин:
1) падіння нейрогенного тонусу артеріол (больові форми шоку);
2) зменшення базального тонусу судин під дією біологічно активних речовин (анафілактичний, панкреатичний шок) або токсичних продуктів (травматичний, турнікетний, інфекційно-токсичний шок)
. Порушення реологічних властивостей крові:
1) синдром внутрішньо судинного дисемінованого зсідання крові (панкреатичний шок);
2) агрегація формених елементів крові (септичний, інфекційно-токсичний шок);
3) згущення крові - гемоконцентрація (ангІдремічний шок).
відбуваються структурні і функціональні зміни у тих органах, за рахунок яких здійснюється централізація кровообігу. Особливо чутливі до шоку легені, нирки, печінка. На початку зміни в них зворотні (нирка в шоці, печінка в шоці, легеня в шоці). При поглибленні гемодинамічних розладів зміни стають незворотними ("шокова легеня", "шокова нирка", "шокова печінка"). Виникає ацидоз і гіпоксичний набряк, агрегація клітин крові, мікротромбози капілярів, внутрішньосудинне згортання крові, погіршуються функції клітинних мембран, внаслідок чого функції органів знижуються і припиняються. Розвивається поліорганна недостатність, яка і визначає прогноз.
* «Шокові органи».
Якщо ОЦК швидко не нормалізується і мікроциркуляція не відновиться – відбуваються структурні і функціональні зміни у тих органах, за рахунок яких здійснюється централізація кровообігу. Особливо чутливі до шоку легені, нирки, печінка. На початку зміни в них зворотні (нирка в шоці, печінка в шоці, легеня в шоці). При поглибленні гемодинамічних розладів зміни стають незворотними ("шокова легеня", "шокова нирка", "шокова печінка"). Виникає ацидоз і гіпоксичний набряк, агрегація клітин крові, мікротромбози капілярів, внутрішньосудинне згортання крові, погіршуються функції клітинних мембран, внаслідок чого функції органів знижуються і припиняються. Розвивається поліорганна недостатність, яка і визначає прогноз.
Причини швидкого зменшення ОЦК – крововтрата, плазмовтрата (опік, перитоніт), дегідратація (діарея, блювання, поліурія, секвестрація рідини у "третьому" просторі – позасудинному міжклітинному), первинні порушення судинного тонусу (при травмах), порушення тканинної циркуляції на ґрунті змін капілярного і посткапілярного опору, шунтування, підвищення проникності капілярів, в результаті чого порушується розподіл крові в тканинах, депонується її частина.
153. Особливості розвитку різних видів шоку: гіповолемічного, кардіогенного, септичного, анафілактичного, травматичного. Crash-синдром.
* Гіповолемічний шок характеризується критичним зниженням переднавантаження на серце .
З точки зору патогенезу можна виділити чотири форми.
1. Геморагічний шок на фоні гострої зовнішньої або внутрішньої кровотечі без значного пошкодження тканин.
2. Гіповолемічний шок на фоні критичного зниження об’єму циркулюючої плазми без гострої крововтрати внаслідок зовнішньої або внутрішньої втрати рідини, або недостатнього надходження рідини до організму.
3. Травматично-геморагічний шок на фоні гострої крововтрати та значних пошкоджень тканин із вивільненням медіаторів внаслідок дії зовнішніх фізичних або хімічних факторів (політравма).
4. Травматично-гіповолемічний шок на фоні критичного зниження об’єму циркулюючої плазми без гострої крововтрати з одночасним значним пошкодженням тканин та вивільненням медіаторів при значних термічних опіках, забоях м’яких тканин.
֍ Гіповолемічний шок характеризується критичним зменшенням тканинної перфузії, викликаним гострим дефіцитом циркулюючої крові, зменшенням венозного притоку до серця і вторинним зниженням серцевого викиду. Основні причини, що викликають зниження об’єму циркулюючої крові: кровотеча, втрата плазматичної рідини і зневоднення .
֍ При більш детальному розгляді патофізіологічних змін в організмі, що виникають внаслідок гіповолемічного шоку, можна виділити декілька фаз .
I фаза — дефіцит об’єму циркулюючої крові. Гострий дефіцит об’єму крові призводить до зменшення венозного притоку до серця та зниження центрального венозного тиску (ЦВТ). Внаслідок цього знижується ударний об’єм серця. Протягом першої години інтерстиціальна рідина спрямовується в капіляри, відповідно знижується обсяг інтерстиціального водного сектора. Загальний обсяг транскапілярного наповнення зростає максимум на 1 л.
II фаза — стимуляція симпатико-адреналової системи. Рефлекторна стимуляція барорецепторов викликає активізацію симпатико-адреналової системи. Порушення її зумовлює підвищення секреції катехоламінів, вміст яких зростає у десятки (норадреналін) та сотні (адреналін) раз. Збільшується симпатичний тонус серця, вен і артеріол, зменшується вагусний вплив на серце. Стимуляція b-адренергічних рецепторів призводить до збільшення скорочувальної здатності міокарда та частоти серцевих скорочень. Стимуляція b-адренергічних рецепторів викликає скорочення селезінки, венозних судин, вазоконстрикцію у шкірі, скелетних м’язах, нирках, призводячи до централізації кровообігу. Цей механізм спрямований на підтримку кровообігу в мозку і серці за рахунок погіршення кровообігу в органах, що іннервуються блукаючим нервом (печінка, підшлункова залоза, кишечник), а також у нирках, шкірі та м’язовій системі. Вазоконстрикція об’ємних судин, що викликає зменшення ємності венозних судин, призводить до диспропорції між обсягом крові і ємністю судинного русла. В короткому інтервалі часу ця реакція є захисною і при швидкій нормалізації об’єму крові настає одужання. Якщо ж дефіцит об’єму циркулюючої крові зберігається, на перший план виступають негативні наслідки тривалої ішемії, за рахунок яких досягається централізація кровообігу.
Кардіогенний шок спостерігається при зниженні насосної функції серцевого м'яза (інфаркт міокарда, міокардит), при тяжких порушеннях серцевого ритму (пароксизмальна тахікардія),
при тампонаді серця (тромбоз порожнин, випіт або кровотеча в навколосерцеву сумку), при масивній емболії легеневої артерії (тромбоемболія легень). Провідним механізмом кардіогенного шоку є зменшення ударного і хвилинного об’єму крові, артеріального тиску і збільшення тиску наповнення серця. Як і при ангідремічному шоку, внаслідок симпатоадренергічної реакції, спостерігається тахікардія, збільшення периферичного опору судин.
Септичний (ендотоксиновий) шок виникає як ускладнення сепсису. Головним фактором, які ушкоджує, є ендотоксини мікроорганізмів. Найбільш частою причиною сепсису є грамнегативні мікроорганізми, а також стрептококи, стафілококи, пневмококи і багато інших.
֍ Провідні патогенетичні ланки септичного шоку:
1) збільшення потреби організму в кисні внаслідок посилення обмінних процесів, тахіпное, тахікардія, ознобом. Потім спостерігається зниження загального периферичного опору судин;
2) зниження оксигенації крові в легенях і недостатній вбирання кисню з крові тканинами. Оксигенація знижена у зв'язку з циркуляторними порушеннями в малому колі, агрегацією тромбоцитів на стінках судин;
3) активація ендотоксинами протеолітичних систем у біологічних рідинах (калікреїн-кінінова, комплемент, фібринолітична).
Анафілактичний шок розвивається внаслідок підвищеної чутливості організму до речовин антигенної природи і нагромадження гістаміну та інших вазоактивних речовин (кініни, серотонін).
При цьому відбувається різке зменшення венозного повернення до серця. Причиною цього є розширення капілярних і ємкісних судин. Скупчення крові в капілярних судинах і венах приводить до зменшення об’єму циркулюючої крові.
Спостерігається і порушення скоротливої діяльності серця. Симпатоадренергічна реакція при цьому не виражена внаслідок порушення судинного тонусу.
Травматичний шок розвивається внаслідок великих пошкоджень тканин. У клініці його розрізняють дві стадії:
1. збудження (еректильну);
2. гальмування (торпідну).
Стадія збудження короткочасна, характеризується порушенням центральної нервової системи внаслідок надходження больових імпульсів з пошкоджених тканин. При цьому розвивається больовий стрес, який проявляється посиленням функцій системи кровообігу, дихання, деяких ендокринних залоз (аденогіпофіза, мозкової і коркової речовини наднирників, нейросекреторних ядер гіпоталамуса) з вивільненням у кров надлишкової кількості кортикотропіну, адреналіну, норадреналіну, вазопресину. Стадія гальмування більш тривала (від декількох годин до доби) і характеризується розвитком у центральній нервовій системі гальмівних процесів. Генералізоване гальмування захоплює і центри життєво важливих функцій (кровообігу, дихання), вони порушуються, внаслідок чого розвивається кисневе голодування. Гіпоксія, у свою чергу, збільшує порушення в серцево-судинному і дихальному центрах.
Розлади гемодинаміки і зовнішнього дихання прогресують - «порочне коло» замикається. Крім нервово-рефлекторних механізмів у виникненні і розвитку травматичного шоку певну роль відіграє також токсемія, обумовлена всмоктуванням у кров продуктів розпаду нежиттєздатних тканин. Останнім часом особливе значення надають так званому ішемічному токсину. Участь токсичних продуктів у патогенезі травматичного шоку доводиться дослідами з “перехресним кровообігом”.
* Crash-синдром.
Краш-синдром (синдром тривалого роздавлювання) - це патологічний процес, який розвивається у потерпілих у результаті тривалого (4-8 г і більше) роздавлювання м'яких тканин кінцівок уламками зруйнованих будинків, споруд, брилами ґрунту при обвалах у шахтах та ін.
֍ У перебігу краш-синдрому розрізняють три періоди:
1) ранній (до 3-х діб) з переважанням явищ шоку;
2) проміжний (з 3-ї до 12-ї доби) з переважанням гострої ниркової недостатності;
3) пізній (з 8-12-ї доби до 1-2 міс), або період видужання, з переважанням місцевих симптомів.
֍ У розвитку краш-синдрому велике значення мають три патогенетичних фактори:
а) больове подразнення;
б) травматична токсемія, обумовлена всмоктуванням токсичних продуктів аутолі-зу тканин з вогнища ураження;
в) плазмо - і крововтрата, пов'язані з набряком і крововиливами в зоні роздавлених або довготривало ішемізованих тканин.
154. Колапс. Етіологія, патогенез.
Колапс — це судинна недостатність, що швидко розвивається і характеризується в першу чергу падінням судинного тонусу, а також гострим зменшенням об'єму циркулюючої крові. При цьому відбувається зменшення припливу венозної крові до серця, зниження серцевого виштовху, падіння артеріального й венозного тиску, порушуються перфузія тканин і обмін речовин, настає гіпоксія головного мозку, пригнічуються життєво важливі функції організму.
Етіологія
Розвивається при масивній крововтраті в результаті швидкого зменшення циркулюючої крові. Колапс може також спостерігатися при гострих захворюваннях внутрішніх органів (перитоніт, гострий панкреатит, дуоденіт, ерозивний гастрит), при захворюваннях серця, які супроводжуються різким і швидким зменшенням ударного об’єму (інфаркт міокарда, порушення серцевого ритму, гострий міокардит або перикардит з нагромадженням випоту в порожнині перікарда).
Патогенез
֍ У патогенезі умовно можна виділити два основних механізми:
1) падіння тонусу артеріол і вен у результаті дії інфекційних, токсичних, фізичних, алергічних і інших факторів безпосередньо на судинну стінку, судинноруховий центр і на судинні рецептори (синокаротидної зони, дуги аорти);
2) швидке зменшення маси циркулюючої крові (крововтрата, плазмовтрата). Зменшення об’єму циркулюючої крові приводить до зниження повернення крові до серця по венах великого кола кровообігу і відповідно серцевого викиду.
При цьому порушується система мікроциркуляції, кров скупчується в капілярах, падає кров'яний тиск, розвивається циркуляторна гіпоксія, метаболічний ацидоз, підвищується проникність судин. Це сприяє переходу води та електролітів із крові в міжклітинний простір, порушуються реологічні властивості крові, виникає гіперкоагуляція крові і патологічна агрегація еритроцитів і тромбоцитів, що створює умови для утворення мікротромбів. При затяжному перебігу колапсу в результаті гіпоксії і порушення обміну речовин звільняються вазоактивні речовини (гістамін, кініни, простагландини) і утворюються тканинні метаболіти –молочна кислота, аденозин і його похідні, які викликають гіпотонію.
Основні прояви патогенезу і загальні прояви колапсу
Ланки
Прояви
Посилення розладів ф-цій ССС
Коронарна недост., зниження ударного і серцевого викиду, гіпоперфузія тканин, венозний застій, перерозподіл кровотоку, капіляротрофічна недостатність;
Порушення функції нервової системи
Загальмованість, апатія, тремтіння пальців рук, судоми, зниж. Нервово-мязової збудливості, розлади свідомості
Розлади газообміну в легенях
Часте поверхневе дихання, гіпоксемія і гіперкапнія крові, відтікаючої від легень
Порушення екскреторної функції нирок
Олігурія, гіперстенурія, гіперазотемія
Порушення функції печінки (при тяжкому перебігу колапсу)
Парціальна або тотальна печінкова недостатність
Розлади в системі крові і гемостазі
Підвищення в’язкості крові, гіповолемія, агрегація тромбоцитів і еритроцитів, тромбоутворення, сладж-синдром
155. Кома. Визначення поняття, класифікація. Роль порушень енергозабезпечення головного мозку, осмотичних розладів, іонного та кислотно-основного гомеостазу в патогенезі коми .
Кома – це патологічний стан, який характеризується глибоким пригніченням функцій центральної нервової системи і проявляється втратою свідомості, відсутністю рефлексів на зовнішні подразники і розладами регуляції життєво важливих функцій організму.
За походженням розрізняють:
1. Коми при первинному ураженні і захворюваннях центральної нервової системи (інсульт, черепно-мозкова травма, запалення, епілепсія,. пухлини головного мозку і його оболонок).
2. Коми при ендокринних захворюваннях, які виникають як при недостатності деяких залоз внутрішньої секреції (діабетична, гіпокортикоїдна, гіпопітуїтарна, гіпотиреоїдна), так і при їх гіперфункції (тиреотоксична, гіпоглікемічна).
3. Токсичні коми спостерігаються при ендогенних (уремія, печінкова недостатність, токсикоінфекції, панкреатит) і екзогенних інтоксикаціях (отруєння алкоголем, барбітуратами, фосфорорганічними та іншими сполуками).
4. Коми, обумовлені порушеннями газообміну при різних видах гіпоксій.
5. Коми, обумовлені втратою електролітів, води та енергетичних речовин.
Провідними патогенетичними ланками в розвитку коми є:
1. Порушення клітинного дихання та обміну енергії в головному мозку (Енергодифіцитний механізм). Основою їх є гіпоксія, анемія, розлади мозкового кровообігу, блокада дихальних ферментів цитотоксичними отрутами, ацидоз (при діабетичній і уремічній комі), дефіцит енергетичних речовин або блокада їх утилізації (голодна і гіпоглікемічна коми). У розвитку гіпоксії мозку мають значення розлади мікроциркуляції. Внаслідок гіпоксії порушується окисне фосфорилювання, зменшується вміст і використання АТФ і креатинфосфату.
2. Порушення синаптичної передачі в центральній нервовій системі. Вони можуть бути пов'язані з:
а) порушенням синтезу, транспорту, депонуванням і секреції нейромедіаторів;
б) витисненням нейромедіаторів псевдомедіаторами;
в) надмірною активацією гальмівних постсинаптичних рецепторів;
г) блокадою збудливих постсинаптичних рецепторів. Цей механізм має велике значення в розвитку печінкової, уремічної і токсичної коми.
3. Порушення балансу електролітів із змінами клітинних потенціалів і процесу поляризації мембран нейронів, а також порушенням осмотичного тиску. = Загальні водно-електролітні порушення. Найбільше значення мають розлади обміну К, Nа, Мg, Са в поєднанні з порушеннями кислотно-основної рівноваги (діабетична, уремічна, хлоргідропенічна, печінкова та ін. коми). Мають значення зміни осмотичного тиску крові й порушення балансу електролітів в організмі.
4. Зміни фізичних властивостей і структури головного мозку і внутрішньочерепних утворів. Патогенетичне значення має набухання і набряк мозку і мозкових оболонок, підвищення внутрішньочерепного тиску, які підсилюють порушення гемодинаміки і ліквородинаміки, обтяжують гіпоксію нервових клітин і пригнічуютьїх фізіологічну активність. Механічне пошкодження клітин мозку має значення при черепно-мозковій травмі, пухлинах, крововиливі в мозок. При окремих видах кому кожний з перерахованих факторів може мати провідне значення, проте частіше діють разом. При глибокій комі розлади регуляції вегетативних функцій приводять додатково до тяжких порушень метаболізму в організмі, у тому числі головному мозку, і створюють “порочне коло” у патогенезі коми.
156. Патогенез різних видів ком: при механічних травмах, метаболічних ком (діабетичних, печінкової, уремічної).
֍ Патогенез ком при механічних травмах:
Зміни фізичних властивостей і структури головного мозку і внутрішньочерепних утворів.
Патогенетичне значення має набухання і набряк мозку і мозкових оболонок, підвищення внутрішньочерепного тиску, які підсилюють порушення гемодинаміки і ліквородинаміки, обтяжують гіпоксію нервових клітин і пригнічуютьїх фізіологічну активність. Механічне пошкодження клітин мозку має значення при черепно-мозковій травмі, пухлинах, крововиливі в мозок. При окремих видах кому кожний з перерахованих факторів може мати провідне значення, проте частіше діють разом. При глибокій комі розлади регуляції вегетативних функцій приводять додатково до тяжких порушень метаболізму в організмі, у тому числі головному мозку, і створюють “порочне коло” у патогенезі коми.
֍ Патогенез діабетичної коми (метаболічна)
(ендогенна кома внаслідок порушення діяльності ендокринної системи) :
Порушення балансу електролітів із зміною клітинних потенціалів і процесів поляризації нейроцитів. Найбільше значення мають розлади обміну К, Nа, Мg, Са в поєднанні з порушеннями кислотно-основної рівноваги.
֍ Патогенез печінкової коми
(ендогенна кома внаслідок порушення діяльності печінки) :
Порушення утворення та виділення медіаторів у синапсах центральної нервової системи. Вони можуть бути пов'язані з: а) порушенням синтезу, транспорту, депонування і секреції нейромедіаторів; б) витісненням нейромедіаторів так званими псевдомедіаторами (хибними медіаторами); в) надмірною активацією гальмівних постсинаптичних рецепторів; г) блокадою збудливих постсинаптичних рецепторів.
֍ Патогенез уремічної коми
(ендогенна кома видільної системи) :
Порушення утворення та виділення медіаторів у синапсах центральної нервової системи. Вони можуть бути пов'язані з: а) порушенням синтезу, транспорту, депонування і секреції нейромедіаторів; б) витісненням нейромедіаторів так званими псевдомедіаторами (хибними медіаторами); в) надмірною активацією гальмівних постсинаптичних рецепторів; г) блокадою збудливих постсинаптичних рецепторів.
* Уремічна (гіперазотемiчна) кома розвивається при гострій або хронічній нирковій недостатності (гломеруллонефрiт, пiєлонефрiт, амiлоїдний нефроз і інші); рясної втрати рідини в результаті неприборканої блювоти або діареї.
2.Розвивається поступово з провісників: з'являються симптоми пригнiчення ЦНС (слабість, головні болі, сонливість, апатія, дратівливість).
3.Спрага, сухість у роті, шкірна сверблячка, на шкірі відкладення кристалів сечовини у вигляді пудри, шкіра суха, блідо-сіра, сліди роздряпин; нудота, блювота кольору «кавової гущавини», пронос; апетит знижений, у роті виразковий стоматит, некротичнi зміни.
4.Запах сечовини у видихуваному повітрі, геморрагiчний синдром.
5.Подих патологічного типу Куссмауля або Чейн-Стокса.
6.Пульс поверхневий, аритмічний, границі серця розширенi, систолiчний шум на верхівці, шум тертя перикарду, шум тертя плеври.
156. Патогенез різних видів ком: при механічних травмах, метаболічних ком(діабетичній, печінкової, уремічної).
За походженням розрізняють:
1. Коми при первинному ураженні і захворюваннях центральної нервової системи (інсульт, черепно-мозкова травма, запалення, епілепсія,. пухлини головного мозку і його оболонок). 2. Коми при ендокринних захворюваннях, які виникають як при недостатності деяких залоз внутрішньої секреції (діабетична, гіпокортикоїдна, гіпопітуїтарна, гіпотиреоїдна), так і при їх гіперфункції (тиреотоксична, гіпоглікемічна). 3. Токсичні коми спостерігаються при ендогенних (уремія, печінкова недостатність, токсикоінфекції, панкреатит) і екзогенних інтоксикаціях (отруєння алкоголем, барбітуратами, фосфорорганічними та іншими сполуками). 4. Коми, обумовлені порушеннями газообміну при різних видах гіпоксій. 5. Коми, обумовлені втратою електролітів, води та енергетичних речовин.
Патогенез при механічних травмах коми. Патогенетичне значення має набухання і набряк мозку і мозкових оболонок, підвищення внутрішньочерепного тиску, які підсилюють порушення гемодинаміки і ліквородинаміки, обтяжують гіпоксію нервових клітин і пригнічують їх фізіол. активність. Механ. пошкодження клітин мозку має значення при черепно-мозковій травмі, пухлинах, крововиливі в мозок. При глибокій комі розлади регуляції вегетативних функцій приводять додатково до тяжких порушень метаболізму в організмі, у тому числі головному мозку, і створюють “порочне коло” у патогенезі коми.
Патогенез метаболічних ком.Порушення балансу електролітів із змінами клітинних потенціалів і процесу поляризації мембран нейронів, а також порушенням осмотичного тиску. Найбільше значення мають розлади обміну К, Nа, Мg, Са в поєднанні з порушеннями кислотно-основної рівноваги (діабетична, уремічна, печінкова коми). Порушення клітинного дихання та обміну енергії в головному мозку. Основою їх є гіпоксія, анемія, розлади мозкового кровообігу, блокада дихальних ферментів цитотоксичними отрутами, ацидоз (при діабетичній і уремічній комі)
157. Значення гіпоглікемічної коми в практиці лікаря –педіатора, її ознаки та міри запобігання.
Гіпоглікемічна кома у дітей розвивається при цукровому діабеті в результаті передозування інсуліну або внаслідок прийому недостатньої кількості вуглеводів після введення інсуліну, а також при спонтайних гіпоглікеміях, які можуть бути функціонального та органічного (пухлини підшлункової залози) походження. Міри запобігання коми –збалансоване спеціалізоване харчування за правильно розрахованим графіком, чітке дозування інсуліну. Кома при гіпоглікемії звичайно виникає раптово, у дітей, хворих на цукровий діабет, в будь-який час доби, а при спонтанній гіпоглікемії дитина впадає в кому частіше вночі. Клінічні прояви гіпоглікемічної коми: гострий початок, рухове збудження, головний біль, підвищена пітливість, слабкість, серцебиття, спостерігаються галюцинації, клонічні та тонічні судоми, тризм жувальної мускулатури, обличчя амімічне, сплутана свідомість, оглушення, що переходить у сопор.
Діагностика гіпоглікемічної коми:
– аглюкозурія;
– рівень кетонових тіл у нормі, ацетон у сечі не визначається;
– КОС не порушений.
158. Особливості шоку у дітей.
Шок — гострий патологічний стан, що виникає через невідповідність перфузії потребам тканин та може призводити до смерті. Характеризується синдромом поліорганної недостатності — зростаючим пригніченням усіх життєвих функцій організму: діяльності центральної та вегетативної нервовової систем, кровообігу, дихання, обміну речовин, функцій печінки та нирок.Метаболічні розлади при шоці у дітей дуже часто призводять до появи аритмій серця. У маленьких дітей ЧСС в більшій мірі визначає величину серцевого викиду, ніж можливість його підтримки за рахунок контрактильности міокарда. Брадиарітмія на тлі метаболічних порушень різко обмежують компенсаторні можливості щодо задоволення зростаючих потреб з постачання кисню і нутрієнтів. Тахіаритмія, що виникають на тлі гіпертермії, гіповолемії, масивного викиду катехоламінів на тлі стресу, порушують продуктивність серця як насоса, істотно збільшують фізіологічну ціну серцевого викиду.
Одна з особливостей перебігу шоку у дітей - невідповідність тяжкості захворювання і тяжкості стану дитини. Можливість підтримувати основні вітальні функції на нормальному рівні зберігається навіть при втрати 25-30% ОЦК. Це відбувається завдяки потужним компенсаторним можливостям дитячого організму. Підвищення загального периферичного судинного опору - єдиний спосіб підтримки адекватної перфузії серця, а також інших життєво важливих органів: головного мозку, нирок. Тому гіпотензія у дітей виникає як пізній і негативний прогностичний ознака.
Шокові стани у дітей супроводжуються пригніченням ретикулоендотеліальної системи, тому в комплекс лікування необхідно включати антибіотики, найважливіший компонент комплексної терапії сепсису.
Кардіогенний - розвивається внаслідок недостатньої функції міокарда; первинно розвивається у дітей з вродженими вадами серця або при гемодинамічно виражених аритміях,вдруге - на тлі гіпоксичних пошкоджень у результаті сепсису, панкреатиту у зв'язку з пригніченням функції міокарда.
159. Особливості гіповолемічного шоку в дітей.
Шок подібного типу може розвинутися при опіковій травмі, гострій хірургічній патології черевної порожнини (кишкова непрохідність, перитоніт), гострого діарейного синдрому різної етіології. У дітей раннього віку найбільш частою причиною гіповолемічного шоку - кишковий є токсикоз з ексикозом інфекційної етіології. Основний етіопатогенетичний фактор шоку - зниження плазматичного об'єму і зменшення повернення венозної крові до серця. Ведучим звеном патогенеза, яке визначає важкість перебігу шоку є значні реологічні і коагулійні порушення в крові. Визначено, що важкі геморагічні порушення виникають при гострому зменшенні плазматичного об'єму та 20 - 30 %. Недосконалість регуляції водно-сольового обміну в дітей молодшого віку сприяє також швидкому і частому розвитку цього виду шоку. Гіповолемічний шок розвивається внаслідок швидкої втрати рідини в організмі. Рідина може втрачатися безпосередньо з кров»яного русла внаслідок кровотечі і це визначається як геморагічний шок, а може через кишечник, шкіру, дихальні шляхи, нирки з переважною втратою води і електролітів і його визначають як ангідремічний шок, тобто до геморагічного шоку зазвичай відносять шок, який розвивається внаслідок втрати (зменшення) ОЦК, а до ангідремічного – коли переважає втрата води і електролітів.
Залежно від тяжкості гіповолемічного шоку, в його перебігу виділяють три стадії, які послідовно один одного змінюють. це
Перша стадія - непрогресуюча (компенсована). Перфузія життєво важливих органів зберігається за рахунок компенсаторних механізмів; як правило, не спостерігається вираженої гіпотензії внаслідок підвищення загального периферичного судинного опору. На цій стадії відсутні порочні круги.
Друга стадія - прогресуюча. Компенсаторні механізми не в змозі забезпечити достатню перфузію, запускаються і прогресують все патогенетичні механізми розвитку шоку.
Третя стадія - стадія незворотних змін. На цій стадії ніякі сучасні протишокові засоби не дозволяють вивести дитину з цього стану. На цій стадії лікарське втручання може повернути на короткий період часу артеріальний тиск і хвилинний об'єм серця до норми, але це не зупиняє руйнівні процеси в організмі. Серед причин незворотності шоку на цій стадії відзначають порушення гомеостазу, яке супроводжується важкими ушкодженнями всіх органів, особливе значення має пошкодження серця.
Критерії діагностики
1. Огляд хворого 2. Вимірювання артеріального тиску, частоти дихання, частоти серцевих скорочень, температури тіла.3. Аускультацію серця.4. Клінічний аналіз крові, сечі.5. Визначення гематокриту.6. Біохімічне обстеження крові.7. Електрокардіографію.8. Поширені біохімічні дослідження крові (коагулограма, протеїнограма, альфа-амілаза, осмолярність та ін.). 9. Дослідження газів крові та кислотно-лужного стану крові. 10. Дослідження імунологічного стану.
Діагноз визначається на основі анамнезу і характерних клінічних симптомів. Цей симптомокомплекс є свідоцтвом дегідратації і гіповолемії, які потребують першочергової корекції. Дегідратацію будь-якого типу визначає універсальний склад симптомів, що відрізняються тільки ступенем їх відбиття -спрага, перебігаюча в анорексію, неспокій, який змінюється млявістю, адинамія, западаннятім»ячка і очних яблук, тахікардія і гіпотензія різного значення, сухість слизових оболонок і шкіра в вигляді "стоячої складки", олігурія або анурія.
До переліку лікувальних заходів входять:
1. Реанімаційні заходи (при необхідності).2. Інгаляція киснем.3. Постановка назогастрального зонду.4. Оральна регідратація.5. Зігрівання дитини.6. Госпіталізація в палату або у відділення інтенсивної терапії.7. Моніторинг артеріального тиску, частоти серцевих скорочень, частоти дихання, пульсоксиметрія. Забезпечення надійного судинного доступу. Визначення центрального венозного тиску. Контроль діурезу. 8. ШВЛ при необхідності. 9. Визначення ступеню компенсації гіповолемічного шоку. 10. Симпатоміметична підтримка кровообігу (допмін, добутрекс).
В лікуванні гіповолемії важлива не тільки корекція втраченого об'єму, але і поновлення мікроциркуляції. Розрахунок об'єму волемічної терапії проводиться на основі вікової фізіологічної потреби в воді, попередніх її втрат, перспіраційних і подовжуючих втрат води.
160. Особливості септичного шоку в дітей.
Септичний шок — невідкладний стан, найтяжчий прояв сепсису, якій виникає при інфекційних, хірургічних, урологічних, акушерсько-гінекологічних хворобах, який проявляється тяжкою гіпотензією і порушеннями системної гемодинаміки. Цей вид шоку відносять до дистрибутивних або перерозподільних. Патофізіологічна відповідь на інфекцію: -активацію нейтрофілів та моноцитів; -вивільнення медіаторів запалення; -дифузну вазодилатацію та збільшення проникності ендотелію; -активацію факторів згортання крові.
У клітинах активується вивільнення цитокінових та нецитокінових медіаторів: -фактор некрозу пухлин-a (TNF-a), -інтерлейкін-1 (IL-1), -інтерлійкін-6 (IL-6).
До кровотоку виділяються медіатори з вазодилатуючою та ендотоксичною дією (простагландини, тромбоксан А2 і оксид азоту), що призводить до системної гіпоперфузії, розвитку синдрому капілярного просочування, мікротромбування, ішемії та функціональної недостатності органів. Слід зазначити, що порушення мікроциркуляції можуть виникати навіть на фоні нормальних показників гемодинаміки.
Найчастішими збудниками позалікарняного септичного шоку у дітей старше 1 міс. без імунодефіциту є: менінгокок, пневмокок, гемофільна паличка типу B.
У дитини гіпертермія, збуджений стан, галюцінації, потім втрата свідомості, інколи судоми, блювотиння. Шкіра бліда з акроціанозом, згодом з’являються петехії. Дихання поверхневе, часте, аритмічне. Тахікардія, зниження артеріального тиску, відставання пульсу на периферічних артеріях. Розвиток олігурії, анурії. Порушення кровообігу у мікроциркуляторному руслі призводить до транслокації екстрацеллюлярної рідини, розвитку інтерстіціального набряку внаслідок чого виникає „шокова легеня”, набряк мозку, „шокова нирка”. Ураження ендотелію судин, мембран лейкоцитів та еритроцитів, агрегація тромбоцитів. Локальні прояви захворювання малоінформативні та визначаються додатковими дослідженнями. Клінічні прояви токсикозу не стабільні, прогресують.
161. Особливості анафілактичного шоку в дітей.
Анафілактичній шок (АШ) – максимально тяжкий прояв алергічної реакції негайного типу. АШ – стан, який виникає гостро і загрожує життю, супроводжується порушенням гемодинаміки, що призводить до недостатності кровообігу та гіпоксії у всіх життєво важливих органах. АШ характеризується швидким розвитком переважно загальних проявів анафілаксії: зниженням артеріального тиску, температури тіла, порушенням функції ЦНС, підвищенням проникності судин, спазмою гладком’язових органів, тощо. АШ виникає після контакту хворого з алергеном, до якого він чутливий: медикаментозні препарати, вакцини, сироватки, харчові продукти, охолодження тіла, яд комах та інші причини.
На частоту і час розвитку АШ впливає шлях введення алергену в організм. У разі парентерального введення алергену АШ частіше. Особливо небезпечний внутрішньовенний шлях введення медикаментозного препарату, хоча АШ цілком можливій при будь-якому шляху застосування лікарських засобів. Виділяють п’ять клінічних форм АШ: асфіктичний, гемодинамічний, абдомінальний, церебральний та змішаний. За типом перебігу АШ може бути: гострий доброякісний, гострий злоякісний, затяжний, рецидивуючий та абортивний.
Діагноз ставиться після збору анамнезу та огляду хворого. Після надання першої медичної допомоги при анафілактичному шоку лікар призначить ряд обстежень. Додаткові дослідження:
- загальний аналіз крові;
- загальний аналіз сечі;
- алергологічне дослідження;
- імуноферментний аналіз;
- рентген грудної клітини, ЕКГ.
Профілактика анафілактичного шоку:
•
ретельно зібраний алергологічний анамнез (особистий і сімейний);
• у хворих із обтяженим алергоанамнезом на сигнальному аркуші історії хвороби
ставлять штамп «алергія» і перелічують лікарські засоби, які викликають
алергію;
• після ін’єкції антибіотика необхідно спостерігати за хворим протягом 20-30
хв;
• медперсонал процедурних, хірургічних, алергологічних та інших кабінетів,
медпунктів повинен бути спеціально підготовленим для надання невідкладної
медичної допомоги при медикаментозному АШ і лікування подібних станів.
В усіх процедурних, хірургічних та інших кабінетах, у медпунктах необхідно мати
протишокову аптечку для надання невідкладної допомоги при АШ.
Після тяжкого перебігу АШ можливий розвиток уражень внутрішніх органів: енцефаліт, міокардит, інфаркт міокарду, нефрит, гепатит, гемопатії, васкуліт тощо. У випадках рецидиву АШ – перебіг завжди тяжкий. У дітей і осіб молодого віку летальність при тяжкій формі істотно нижча, при цьому до 70 % її випадків обумовлено гострою дихальною недостатністю внаслідок набряку слизових оболонок верхніх дихальних шляхів, гіперсекреції, бронхо- і ларингоспазму
При деяких гельмінтозах може виникнути анафілактичний шок. У більшості випадків анафілактичний шок з тяжким перебігом і високою летальністю виникав у дітей до 15 років під час міграційної фази аскарид. Аналогічні випадки описані й в окремих хворих на трихінельоз, стронгілоїдоз, вухереріоз, цистицеркоз. Часто розвивається цей вид шоку при розривах ехінококових і альвеококових міхурів унаслідок одномоментного масивного надходження продуктів життєдіяльності гельмінта в кров.
Симптоми анафілактичного шоку в дітей: відчуття страху, занепокоєння, головний біль, шкірні висипання, пирхавка в горлі, тахікардія, біль у животі (не завжди). Швидкість проявів залежить від способу попадання алергену в організм. При внутрішньовенному введенні анафілактична реакція виникає миттєво. При прийомі їжі – протягом декількох годин. Спрогнозувати розвиток анафілаксії неможливо, тож лікарі рекомендують в домашній аптечці завжди мати протиалергійні препарати.
Спочатку повинен бути локалізований або тотальний свербіж шкіри, паління язика, долоней, підошви ніг, потім генералізована кропивниця, набряк Квінке. Швидке падіння артеріального тиску навіть до його відсутності, надмірна тахікардія або брадикардія, порушення свідомості, різка блідість шкіри та слизових оболонок, серцебиття до 200 і більше скорочень на 1 хв. або брадикардія, глухі тони. Порушення дихання, бронхоспазм, набряк гортані, можливі судоми із зупинкою дихання. Може розвинутися синдром "гострого неефективного серця", макульозно-папульозний висип (з'являється при експозиції алергену від 30 хв. і більше).