45.Обмін циклічної амінокислоти триптофану та його спадкові ензимопатії.
- серотоніновий шлях, що становить в кількісному відношенні приблизно 1% загальної кількості триптофану в організмі; Включення триптофану в серотоніновий шлях починається з гідроксилювання амінокислоти до 5-окситриптофану, який після декарбоксилування перетворюється на серотонін. В організмі людини серотонін підлягає окиснювальному дезамінуванню з утворенням оксііндолацетатної кислоти, яка виділяється з сечею. Екскреція оксііндолацетату значно збільшена при карциноїдному синдромі, коли за серотоніновим шляхом перетворюється до 60 % триптофану.
- кінуреніновий шях, за яким метаболізується понад 95 % ендогенного триптофану. Катаболізм триптофану кінуреніновим шляхом починається з окиснення триптофану при дії гемвмісного ферменту триптофанпіролази до формілкінуреніну, який після відщеплення мурашиної кислоти перетворюється на кінуренін та 3-оксикінуренін. Подальші перетворення 3-оксикінуреніну пов'язані з дією ПАЛФ-залежного ферменту кінуренінази, яка розщеплює його до аланіну та 3-оксіантранілової кислоти. Остання після складних багатоступеневих перетворень призводить до хінолінової кислоти — попередника в синтезі нікотинаміду в формі коферменту НАД.
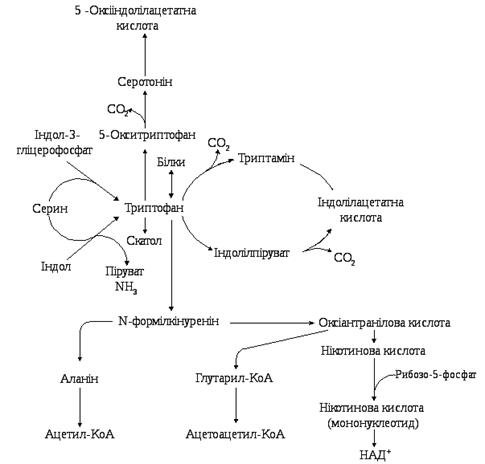
Порушення метаболізму Основний шлях метаболізму триптофану приводить до синтезу аміду нікотинової кислоти, що відіграє дуже важливу роль у життєдіяльності організму, будучи простетичною групою низки окисних ферментів — нікотинамідаденіндинуклеотиду (НАД) і його відновленої форми нікотинамідаденін-динуклеотидфосфату (НАДФ). Тому при недостатності нікотинової кислоти та її аміду порушуються багато обмінних реакцій, а при значному дефіциті цих речовин розвивається пелагра[en]. Порушення обміну триптофану може проявитися також у зміні кількості утвореного з нього серотоніну[8]. Ураження нирок викликається підвищеною екскрецією метаболітів триптофану. Вроджена відсутність триптофан-піролази (ферменту, що окисляє триптофан) призводить до розумової відсталості. Порушення обміну триптофану у людини може сигналізувати про низку захворювань, як то: туберкульоз, рак, діабет.
Гіпертриптофанемія
Синдром Хартнапа— спадкове захворювання, що характеризується пелагроподібним дерматитом, періодичними нападами мозочкової атаксії, гіпераміноацидурією та підвищеною нирковою екскрецією індолацетату (α-N-[індол-3-ацетил]глутамін) і триптофану. У деяких дітей виявляється розумова недостатність. Успадковується за автосомно-рецесивною ознакою. Синдром Хартнапа зумовлений порушенням транспорту триптофану на рівні клітин слизової оболонки кишківника і проксимального відділу ниркових канальців. Це приводить до підвищеного виділення триптофану зі сечею і зниження його абсорбції у кишківнику. Нагромадження триптофану у кишківнику сприяє розкладанню його бактеріями й утворенню великої кількості індольних сполук, що всмоктуються у кров і у більшій кількості виділяються зі сечею. Підвищення вмісту цих сполук у крові, імовірно, обумовлює підвищену чутливість шкірних покривів до дії сонячних променів. Виведення ж значної кількості індикану нирками, у зв'язку з ушкодженням проксимальних канальців, викликає гіпераміноацидурію.Характерним є пелагроподобне ураженням шкіри, що характеризується появою на відкритих ділянках тіла гіперемії, лущення. Ці зміни підсилюються літньої пори під впливом сонячних променів. На шкірі, найчастіше в ділянці носа, виявляються атрофічні ділянки як результат світлової травми, тому хворому рекомендують уникати інсоляції.При лікуванні застосовують вітаміни В6, РР і B1. У періоди загострення — обмеження вживання білків, проведення цукрово-фруктових днів, антибактеріальні препарати для зменшення утворення індольних похідних за рахунок бактеріального розкладання триптофану.
Синдром Тада — спадкове захворювання з автосомно-рецесивним типом успадковування. Вперше описаний К. Тада в 1963 році. Під час цього захворювання спостерігається недостатність ферменту триптофанпіролідази, який каталізує перетворення триптофану в кінуренін. Клінічно виявляється подібність зі синдромом Хартнапа, відрізняється від нього більшою виразністю враження ЦНС і нанізмом. Лабораторні дані також ідентичні, за винятком рівня триптофану в крові, що при даному синдромі завжди підвищений. Порушення пов'язані з ендогенним дефіцитом нікотинової кислоти й надлишком індольних сполук. Синдром Тада спричиняє глибоку розумову відсталість, нанізм та мозочкову атаксію.
Індиканурія Захворювання спричиняється порушенням всмоктування триптофану в кишківнику з утворенням надлишкової кількості індолу, який всмоктується, окислюється, сульфатується і виділяється у формі індикану. Останній окислюється під дією повітря до голубого індикану, який забарвлює пелюшки хворої дитини в синій колір. При індиканурії спостерігається гіперкальциємія, нефрокальциноз, періодична гіпертермія.
Синдром Прайса Генетичний дефект кінуренінгідроксилази. Спостерігається надлишкове виділення зі сечею кінуреніну через заблокованість ферменту. Головний прояв синдрому Прайса — склеродермія.
Основи молекулярної біології